6. STRIPPING
VOLTAMMETRY
6a.
ANODIC-STRIPPING ANALYSIS
Principles of the method
Anodic-stripping analysis is the most sensitive of all commonly used electroanalytical techniques. Analyses can be performed at the trace level and are applicable to solutions containing metal ions in the concentration range of 10-6 - 10-12 M. Other advantageous features of stripping voltammetry include the capability for simultaneous multielement determination and relatively inexpensive instrumentation compared to that required for the spectroscopic techniques.
Three types of working electrodes can be used:
(a) the classical hanging mercury-drop electrode (HMDE),
(b) the thin-film mercury electrode (TFME),
(c) solid electrodes (gold, silver, surface-modified carbon).
The sequence of steps of the anodic stripping analysis is described below.
Step
1. The electrodeposition step
The metal ions Mn+ of interest are deposited (preconcentrated) electrochemically into or onto the surface of an electrode (usually a mercury film electrode or a hanging mercury-drop electrode), in the form of amalgam, M(Hg):
![]()
A short-time electrolysis (30 sec to 5 min) in a stirred solution and at a potential suitable for the reduction of the ions of interest (E -E1/2 about -200 mV) may result under proper conditions in a fairly concentrated amalgam. This step is called the electrodeposition step. The concentration of the metal ion in the film depends on concentration of Mn+ in solution, time of electrolysis and rate of stirring. Since the electrodeposition is carried out on small electrodes, the amount of material deposited into it usually does not change significantly the concentration of the metal ions Mn+ in the solution. Step 1 is only an intermediate step and there is no need to know the concentration reached in the amalgam. However, it can be estimated.
The enrichment of the metal M in the amalgam in respect to the initial concentration of Mn+, CMn+, is estimated in stirred solutions, using Nernst simplified model (eq.3 in section "Coulometric titrations").
![]()
For typical laboratory conditions the Nernstian layer
thickness in well-stirred solutions is about 20 ![]() .
.
As a result of electrolysis for a time t, the concentration of M in the amalgam, CM(Hg), is
![]()
where A and V are the area and the volume of the mercury electrode.
The enrichment of the metal in the amalgam, using D = 10-5 cm2/s, is
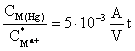
The enrichment factor at HMDE
For a hanging mercury-drop electrode A/V = 3/r, and for r ~0.03 cm it is 100 cm-1, thus
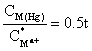
For a 100 s deposition time, the enrichment factor is 50.
The enrichment factor at TFME
The TFME consists of a thin mercury film coated on glassy carbon. A/V for this electrode depends solely on the thickness of the film. Typical dimensions for a thin-film mercury electrode are: A = 0.2 cm2 and film thickness about 10-5 cm. The value of A/V for TFME is 105, which is 1000 times that of HMDE.
![]()
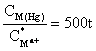
As a result of a 100 sec electrolysis, the concentration of the metal in the amalgam will be ~5·104 times larger than that of the metal in the solution.
The enrichment factor for a hanging mercury-drop electrode is 1000 times smaller than that for a thin mercury film. Nevertheless, substantial increase in concentration is achieved also with this type of electrode for electrolysis time t larger than 100 sec.
The degree
of decrease of concentration in solution as result of the electrodeposition
step is calculated for a mercury film electrode for the following conditions:
the area of the film, A = 0.2 cm2; the initial concentration of the
metal ions in the solution, CMn+ = 10-8 M; the
volume of the tested solution is 20 ml; the time of deposition is 100 s; the
Nernstian layer thickness during the deposition step is 20 ![]() . At the end of the electrolysis step the number of moles of
the metal electrolyzed in the film, CM(Hg) = 10-12 mole
(verify this value!). Thus, the decrease of concentration of Mn+ in
solution after the electrolysis is negligible (0.5 %).
. At the end of the electrolysis step the number of moles of
the metal electrolyzed in the film, CM(Hg) = 10-12 mole
(verify this value!). Thus, the decrease of concentration of Mn+ in
solution after the electrolysis is negligible (0.5 %).
Step
2. Rest period
After a predetermined time, the stirring of the solution is turned off. The solution is allowed to become quiescent and the concentration of the metal in the amalgam - to reach uniformity. The rest period extends for about 30 sec, during which the applied potential remains unchanged, thus ensuring that no reoxidation of the metal by traces of oxygen takes place. During the rest period the electrodeposition current decreases (Explain why).
Step 3. Stripping
After the preconcentration step, the deposited metal M is oxidized ("stripped") from the mercury electrode back into the solution by oxidation to the ionic form under conditions of diffusion control, using one of the voltammetric methods:
![]()
The anodic diffusion current is used to determine the concentration of the metal in the amalgam, which is proportional to time of electrolysis, stirring rate and concentration of Mn+ in solution.
Cleanliness requirement for glassware
The glassware you will use is especially cleaned for this highly sensitive determination. Use only glassware on which the type of chemical and its concentration are marked. Do not interchange glassware. In order to minimize contamination, the amount of glassware should be reduced as much as possible. For example, the sample will be introduced into the cell not with a pipette, but directly from the sample bottle by weighing.
Sample preservation
Drinking water samples should be preserved by adding 10 ml of 1 M HNO3 to each liter of sample immediately after collection. Acidification to a pH between 1 and 2 is an essential first step in water sampling: a) it arrests biological growth; b) it affects the release of trace metals from organic components (humic and fulvic acids) and c) it reduces metal loss caused by adsorption on the container wall.
Sample pretreatment
The presence of surface-active compounds in the samples consists a major problem in the use of solid electrodes. Surfactants compete with the analyte on the electrode surface for active adsorption sites, causing distortion, lower sensitivity and instability of the signal of the analyte. The hanging mercury-drop electrode, owing to its renewable surface, is inherently less sensitive than solid electrodes to surfactants.
Severe peak distortions are overcome with different methods: UV irradiation2 of the sample, coating the electrode with permselective film or membrane that prevents the surfactant from reaching the electrode surface3, addition of fumed silica to the analyte4, wet digestion and others.
Exp.1.
Determination of Pb2+ and Cd2+ in drinking water,
using hanging mercury-drop electrode
Chemicals 1. 1.00·10-4 M Pb(NO3)2
in 10 mM HCl
2. 1.00·10-4 M CdCl2 in 10 mM HCl
3. 10 mM HCl – supporting electrolyte
4. 1 M HNO3
Electrodes: working - HMDE
reference - Silver wire (1.5 mm diameter)
Basic procedure for the anodic-stripping analysis
Prepare fresh solutions of Pb2+ and Cd2+, 10-6 M each, in 10 mM HCl. For drinking water samples: collect tap water into a clean plastic vessel, add 10 ml of 1 M HNO3 to each liter of water, as needed for sample preservation.
Pipet into the cell 5.00 ml of the supporting electrolyte and 0.05 ml of one of the 10-6 M fresh prepared solutions of the metal. Perform an anodic-stripping analysis under the following conditions:
|
Method – Voltammetric analysis: SW |
|
|
Deaeration step (bubbling N2) |
t = 120 s |
|
Preconcentration step (Deposition): |
E = -900 mV t = 60 s |
|
Rest step (Equilibration): |
t = 5 s |
|
Anodic dissolution step: |
Potential scan = from -900 to -200 mV step duration = 0.1 s step amplitude = 5 mV pulse amplitude = 25 mV |
|
Stirring rate |
800 rpm |
Test of reproducibility. Repeat several times the basic procedure in the same solution. Report the average and the standard deviation of the slope.
Effect of electrolysis time. Repeat the stripping procedure with different electrolysis times: from 30 to 120 minutes. Note that the signal is linearly changed with the time.
Calibration curve. A calibration curve for the analyzed metal is constructed for two reasons: (a) to enable rough estimation of the concentration of the metal in the unknowns by comparing peak heights, and (b) to test if linearity between analytical signal and concentration is obtained in the concentration range if interest. This is a basic condition for applying the standard-additions method, used for the analytical determination in this experiment.
Insert into the cell 5.00 ml of the supporting electrolyte. Record a stripping curve of the blank following the basic procedure. Repeat the experiment with five subsequent additions of 0.05 ml of the 10-6 M Pb2+ solution. Plot the calibration curve.
Determination of Cd2+ and Pb2+ in
drinking water
Carry out at least three stripping voltammograms of a drinking water sample. On the basis of your acquired experience make a rough estimation of the concentration of the analyte. Make an appropriate standard addition. Run a new voltammogram. If your guess was successful, repeat the voltammogram. If not, make a new guess.
Report the concentration of the analyte in mol/l and ppb. Compare your results with the official permissible levels.
Exp. 2.
Determination of Pb2+ in drinking water, using the silver
rotating-disk electrode
The Rotating-Disk Electrode (RDE)
One of the best methods of obtaining efficient mass transport in a highly reproducible manner is by the use of the rotating-disk electrode. The RDE consists of a cylindrical metal rod embedded in a larger cylindrical plastic (e.g., Teflon) holder. The electrode is cut and polished flush with its holder, so that only the bottom end of the metal cylinder is exposed to the solution.
The configuration of a rotating-disk electrode is shown schematically in Fig.6-1. The most important feature of the RDE is that it acts as a “uniformly accessible surface”, which means that the rate of mass transport to the surface is uniform. This is by no means self evident, considering that the linear velocity of points on the surface increases with their distance from the center of rotation. The other important property of the RDE is that flow of the solution around it is laminar up to rather high rotation rates.

Fig.6-1 The rotating-disk electrode. The electrode and the insulated material are marked by filled and clear areas, respectively.
Since the
flow is laminar, it is possible to calculate rigorously the rate of mass transport.
The rotating-disk electrode was developed following the mathematical solution
given by Levich of the hydrodynamic equations describing the rate of transfer
of substance in solution to a rotating disk surface, in terms of the angular
velocity of rotation ![]() (
(![]() , N in rps), the diffusion coefficient D, the concentration C0
of the substance and the kinematic viscosity
, N in rps), the diffusion coefficient D, the concentration C0
of the substance and the kinematic viscosity ![]() of the solution. For
the case when the reaction is relatively fast and the current is determined by
mass transport, the corresponding equation for the limiting current density
of the solution. For
the case when the reaction is relatively fast and the current is determined by
mass transport, the corresponding equation for the limiting current density ![]() , developed by Levich, is:
, developed by Levich, is:
![]()
where ![]() is in A/cm2,
D in cm2/s,
is in A/cm2,
D in cm2/s, ![]() in cm2/s ,
in cm2/s ,![]() in rad/s and C0
in mol/cm3. In such a case the limiting current is independent of
potential over a wide range. This range of potential is limited at one end by
the reversible potential and a small overpotential needed to drive even a very
fast reaction to mass-transport limitation and at the other end by another
reaction which may take place, usually the evolution or oxygen or hydrogen in
aqueous solutions.
in rad/s and C0
in mol/cm3. In such a case the limiting current is independent of
potential over a wide range. This range of potential is limited at one end by
the reversible potential and a small overpotential needed to drive even a very
fast reaction to mass-transport limitation and at the other end by another
reaction which may take place, usually the evolution or oxygen or hydrogen in
aqueous solutions.
Experimental procedure
The silver rotating-disk electrode4 is well suited for the determination of lead in ppb and sub-ppb concentrations.
A two-electrode cell configuration is used (cf., Fig.6-2). The working electrode is a silver rotating disk. A silver wire acts as a combined counter/quasi-reference electrode. The potential of the silver wire is stabilized by the presence of chloride ions in working solution.
Handling of the silver disk electrode. The silver disk electrode can be used over hundreds of measurements without any pretreatment, provided the surface-active agents in the samples are destroyed. The proper functioning of the electrode is judged according to two criteria:
(a) the reproducibility of the anodic stripping voltammograms;
(b) the degree of linearity between the analytical signal and the concentration.
If the criteria are not fulfilled to our satisfaction, the electrode should be polished with 0.05 mm alumina powder, rinsed thoroughly with distilled water, sonicated for 3 min for removal of alumina particles and rinsed again with distilled water.

Fig.6-2 Electrochemical cell for anodic-stripping voltammetry.
Chemicals 1. 1.00·10-4 M Pb(NO3)2
in 10 mM HCl
2. 1 M HNO3
3. Combined 10 mM HNO3 and 10 mM NaCl supporting electrolyte
4. Digestion solution containing 2 M HNO3 and 2 M H2SO4
5. 0.05 ![]() alumina powder
alumina powder
Electrodes: working - Silver disk electrode (area about 0.07 cm2)
reference - Silver wire (1.5 mm diameter)
Basic procedure for anodic-stripping voltammogram
Since the work is performed in trace concentrations of analyte (about 10-8 M), care should be taken in the use of utensils: for each concentration use only the vessels marked respectively.
1. In a 100 ml volumetric flask prepare solution of 1.00·10-6 M Pb(NO3)2 in 10 mM HNO3 from the 1.00·10-4 M Pb(NO3)2 stock solution.
2. Pipet into the cell 5.00 ml of the combined 10 mM HNO3 and 10 mM NaCl supporting electrolyte and 0.200 ml of the 10-6 M solution of Pb2+. Start the stirrer.
3. Perform an anodic-stripping analysis at a rotation rate of 3500 rpm under the following conditions:
|
Method – Voltammetric analysis: SW |
|
|
Pretreatment step (Conditioning): |
E = -40 mV |
|
|
t = 10 s |
|
Preconcentration step (Deposition): |
E = -700 mV |
|
|
t = 30 s |
|
Rest step (Equilibration): |
t = 3 s |
|
Anodic dissolution step: |
Potential scan = from -700 to -40 mV |
|
|
Frequency = 25 Hz |
|
|
Step potential = 25 mV |
|
|
Amplitude = 5 mV |
The analytical signal can be quantified by: a) the stripping peak current, b) the stripping peak area, and c) the average slope at the two inflection points of the peak. Use the slope for quantitative evaluation of analyte, since this parameter yields the best linear response vs. concentration.
3. Precision. The precision of an analytical method is the degree of mutual agreement among results obtained under identical conditions. Statistically, the precision is expressed as the relative standard deviation of a set of measurements.
Run five subsequent voltammograms and calculate the average and the relative standard deviation of the slope.
4. Effect of rotation rate. Repeat
the stripping procedure for different rotation rates (2000 - 8000 rpm). Plot
the analytical signal as function of the square root of the rate of rotation.
5. Effect of electrolysis time. Repeat the stripping procedure with different electrolysis times: from 15 to 120 seconds.
6. Background correction. Traces of impurities in the reagents contribute to the background and should be taken into account. Pipet into the cell 5.00 ml of the combined 10 mM HNO3 and 10 mM NaCl solution and run a stripping voltammogram in the supporting electrolyte only. Note the value of the analytical signal of lead in the background (the slope) and use it later to correct the calibration curves.
7. Calibration curve. A calibration curve in the range of 0 to 8·10-8 M of Pb2+ is constructed for two reasons: (a) to enable rough estimation of the concentration of Pb2+ in the unknowns by comparing peak heights, and (b) to test if linearity between analytical signal and concentration is obtained in the concentration range if interest. This is a basic condition for applying the standard-additions method, used for the analytical determination in this experiment.
Insert into the cell 5.00 ml of the combined supporting electrolyte. Record a stripping curve of the blank by following the basic procedure, but increase the deposition time to 60 s. Repeat the experiment with five subsequent additions of 0.1 ml of the 1.00·10-6 M Pb2+ solution. Plot the calibration curve.
Determination of Pb2+ in drinking water
1. Collect a tap water sample as discussed in “Sample preservation”. For preservation of sample and proper functioning of the reference electrode, add 1 ml of the 1 M HNO3 to 100 ml of tap water. If the analysis is carried out immediately after the sample collection, the acidification step can be omitted.
2. Pretreatment of samples. Water samples are pretreated in order to overcome interferences due to surface-active compounds. The procedure applied consists of digestion with nitric and sulfuric acids and heating to 650°C. Nitric acid destroys organic materials and sulfuric acid supplies sulfate ions to ensure that the lead ions form a temperature-stable lead sulfate.
The efficiency of the pretreatment is tested by comparing the lead content in pretreated and unpretreated synthetic solutions.
Digestion of unknowns, blank and control sample
(a) Unknown: 50.00 ml drinking water, acidified for preservation.
(b) Blank: 50.00 ml Type I Reagent Grade Water, acidified as the unknown.
(c) Control sample: 50.00 ml 2·10-8 M Pb2+, acidified as the unknown.
Perform the digestion of the duplicates of: (a) the unknown drinking water sample, (b) blank, and (c) the synthetic control sample (the latter is used to test if lead is lost during the digestion procedure), according the following procedure: insert 5.00 ml of the sample to be digested into a quartz beaker (quartz is stable at high temperatures, and the leaching of lead from quartz is considerably less than from Pyrex glass), add 2 ml of the digestion mixture (2 M HNO3 + 2 M H2SO4) to each beaker.
Transfer the beakers to a muffle furnace and keep at 300°C for about an hour to evaporate gently the solution to dryness. Then heat the furnace to 650°C and keep the beakers for additional 20 minutes. Allow the beakers to cool to room temperature, and add 5.00 ml of the combined 10 mM HNO3 and 10 mM NaCl supporting electrolyte to each one of them.
3. Analysis of the pretreated samples. Add 4.00 ml of the undigested 2·10-8 M Pb2+ control sample (c) to the electrochemical cell. Perform twice the basic stripping procedure with a deposition time of 60 s. Run another voltammogram with a new 4.00 ml aliquot of the control solution to exclude a possibility of contamination of the cell. Verify that the reproducibility of the three measurements is satisfactory. Run a blank stripping voltammogram using 4.00 ml of the combined supporting electrolyte.
Repeat the above step using the digested 2·10-8 M Pb2+ solution – sample (c). Run the voltammogram of the digested blank – sample (b). Calculate the relative difference of the signals of the treated and the untreated solutions each one corrected for the respective blanks.
For the unknown samples (a) and the blank (b) perform a quantitative determination based on standard-additions method. Run stripping voltammogram of the sample. Make a rough estimation of the lead concentration by comparing the analytical signal of the unknown with those of the calibration curve. If the agreement between the duplicates is satisfactory, perform the standard-additions procedure on one of the duplicates only. Keep the following rules in respect with the standard additions: (a) the volume of a single standard addition should be smaller than 5% of the volume of the solution in the cell; (b) the increase in concentration with each addition should be lower than 40% of the concentration of lead, roughly estimated by consulting the calibration curve, recorded under identical conditions. Calculate the concentration of lead in the digested unknown sample and in the digested blank.
Calculate the concentration of lead in the drinking water sample. Report the concentration of the analyte in mol/l and ppb. Compare your results with the official permissible levels.
Exp. 3.
Measurement of contamination of water by Pb2+ as a result of
contact with ceramic glazes and crystal glasses,
using the silver rotating-disk electrode
Lead compounds have been and are still used in the manufacture of ceramic glazes and crystal glasses.
Certain compounds of lead, particularly brightly colored lead oxides, have been used for leaded glasses and leaded glazes on ceramics for thousands of years. The addition of lead improves the appearance and cutting properties of crystal glass. Small amounts of lead are also present in optical glass. The major application of leaded glass is in television screens and computer monitors, and serves to protect viewers from the harmful X-rays generated by these appliances. Lead-containing glazes are used in some pottery, tiles and tableware.
Crystal glasses contain up to 30 % lead oxide, PbO. The European definition of lead crystal as glass that contains a minimum of 24% lead oxide is a feature that once contributed to its reputation for quality. The purpose of lead oxide is to increase the density of glass, which in turn, increases the refractive properties. When it is cut and polished, it creates more colors and sparkle than regular glass. On contact with water or other solutions, the lead is slowly released in trace amounts, especially in acidic solutions.
To read more about the
properties of lead Go
to
http://www.ldaint.org/factbook/factbookch1.htm
http://ianrpubs.unl.edu/water/g1333.htm
Electrodes: working - Silver disk electrode (area about 0.07 cm2)
reference
- Silver wire (1.5 mm diameter)
Chemicals 1. 1.00·10-4 M Pb(NO3)2
in 10 mM HCl
2. 1 M HNO3
3. Combined 10 mM HNO3 and 10 mM NaCl supporting electrolyte
4.
0.05 ![]() alumina powder
alumina powder
Experimental procedure
The same configuration of the electrochemical cell is used (see Exp. 2).
For the preparation of the sample, heat about 100 ml of deionized water in a glass beaker until the water boils, add 1 ml of 10 mM HCl (pH 4). Transfer the hot water to a glazed ceramic vessel or to a crystal glass, wait for about 20 min. Use this solution for determination of the degree of contamination by Pb2+.
Follow the basic procedure of the anodic stripping (see Exp. 2) for recording voltammograms for blank and analyte solutions.
Perform a quantitative determination using the standard-addition method. Consult with the instructor about the details, concerning the volume of the sample and the standard additions.
Estimate the values of the analytical signal from the voltammograms. Plot the analytical signal corrected for background vs. concentration of the metal in the cell. Report the concentration of the metal in the sample in mol/l and ppm.
References
1. C. M. G. Van den Berg, Anal. Chim. Acta, 1991, 250, 265.
2. J Jarbini, and W. R Heineman, Anal. Chim. Acta, 1986, 186, 11.
3. A. Economou and P. R. Fielden, Analyst, 1993, 118, 1399.
4. M. Brand, I. Eshkenazi and E. Kirowa-Eisner, Anal. Chem., 66, 4660(1997).
5. E. Kirowa-Eisner, M. Brand and D. Tzur, Anal. Chim. Acta, 385, 325(1999).
Recommended
Literature
1. D. A. Skoog, Principles of Instrumental Analysis.
2. D. A. Skoog and D. M. West, Principles of Instrumental Analysis.
3. D. A. Skoog and J. J. Leary, Instrumental Analysis.
4. A. J. Bard and L. R. Faulkner, Electroanalytical Chemistry.
6b. ADSORPTIVE
STRIPPING VOLTAMMETRY
Adsorptive stripping voltammetry (AdSV) is similar to anodic-stripping voltammetry (ASV). The method consists of two main steps, in analogy to ASV:
(a) Accumulation step, in which the metal ion is accumulated on the electrode as an adsorbed metal complex.
A surface-active complexing agent is added in the solution (at a concentration not greater than 1 mM). The ligand forms an adsorbed layer on the electrode surface (usually on a hanging mercury-drop electrode) as soon as it is introduced into the solution. The analyte (at nanomolar or sub-nanomolar concentration) reacts with the adsorbed ligand to form an adsorbed complex.
![]()
This process is carried out in stirred solution and under potential control. The analyte reaches the electrode by convection (as in the preconcentration step of anodic-stripping voltammetry).
(b) Stripping the adsorbed metal ion, which is realized by electroreduction of the adsorbed complex.
Square-wave voltammetry is used in this step. The amount of adsorbed metal is determined from the value of the peak current or from the sum of the slopes at the inflection points of the peak.
The ligands used in AdSV are
surface-active compounds or are able to interact chemically with mercury (the
presence of ![]() -electrons benefit the adsorption process and the presence of
S donors helps the chemisorption process). The kinetics of the complex
formation must be fast.
-electrons benefit the adsorption process and the presence of
S donors helps the chemisorption process). The kinetics of the complex
formation must be fast.
In this experiment the ligands are dimethylglyoxime (DMG) and 8-hydroxyquinoline (oxine). In some applications the use of mixture of ligands enables simultaneous determination of up to six trace metals.
The detection limit of AdSV depends on the chemistry of the system and can be as low as 10-12 M.
Adsorptive stripping suffers from interferences resulting from competitive adsorption of surface-active substances present in the analyte. The most common methods for destroying organic matter in the samples are: addition of fumed silica (adsorbs the surface-active substances), ultraviolet irradiation, wet and dry ashing.
Cleanliness of glassware
The glassware used is especially cleaned for this highly sensitive analysis. Prior to use the glassware is immersed for a few hours in a liquid cleaner, followed by immersion in 1:1 aqueous HNO3 and then rinsed with copious amounts of deionized water.
It is recommended to use dedicated vessels for the various analytes and for different concentrations.
In order to minimize contamination, the
amount of glassware and plastic ware should be reduced as much as possible.
Exp. 1. Simultaneous determination of
traces of heavy metals
released from domestic utensils.
This experiment follows the work of C. M. G. van den Berg et al1. Van den Berg demonstrated the advantages of using mixture of ligands in AdSV. With DMG (dimethylglioxime) as ligand only Ni and Co are determined by adsorptive stripping voltammetry 2,3. Addition of oxine (8-hydroxyquinoline) enables simultaneous determination of several metals: Cu, Pb, Cd, Ni, Co and Zn. The concentration range depends on the species present and is roughly from sub-nanomolar up to about 50 nM. At higher concentrations of the metals deviation from linearity may be observed. Thus, an excess of one of the analytes may interfere with the analysis. For this reason, drinking tap water, which normally contains micromolar concentration of Zn, cannot be analyzed by using mixture of ligands, the content of Ni and Co in tap water can be determined using DMG only.
(In this experiment the HEPES buffer has been replaced with an ammonia buffer).
The method of standard additions is used for the quantitative determination of the metal concentrations.
Chemicals 1. 0.5 M ammonia buffer pH 9.24
2. 0.01 M DMG in 96% ethanol
3. 0.1 M oxine in 0.15 M HCl (stock solution, stored in refrigerator)
4. 1 mM oxine (freshly prepared monthly)
5. 10 mM
HCl
6. 0.1 M NaCl
7. 0.1 mM Cu2+ in 10 mM HCl
8. 0.1 mM Pb2+ in 10 mM HCl
9. 0.1 mM Cd2+ in 10 mM HCl
10. 1.7 mM (100 ppm) Ni2+ in 10 mM HCl
11. 1.7 mM (100 ppm) Co2+ in 10 mM HCl
12. 0.05 mM Zn2+ in 10 mM HCl
Electrodes: working - Hanging mercury-drop electrode (HMDE)
reference - Ag/AgCl/1 M KCl
counter
- Pt wire
Basic conditions for adsorptive stripping voltammetry
Deaeration step (bubbling N2)
|
t = 120 s |
|
Accumulation (adsorption ) step |
E = -200 mV t = 30 s |
|
Rest step |
t = 10 s |
|
Reduction (stripping) step |
technique – SWV E = from -200 to -1400 mV step duration = 0.1 s step amplitude = 5 mV pulse
amplitude = 25 mV |
|
Stirring rate |
800 rpm |
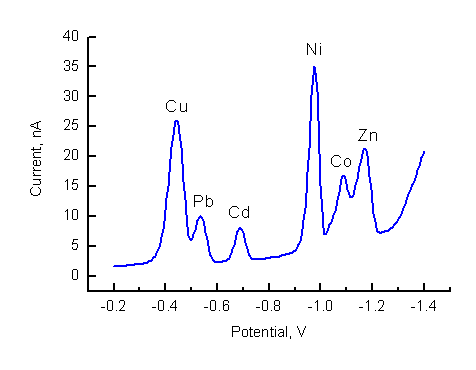
Fig.6-3 Simultaneous determination of 15 nM Cu2+, 15 nM Pb2+, 15 nM Cd2+, 11 nM Ni2+, 11 nM Co2+ and 15 nM Zn2+
in 82 mM ammonia buffer pH 9.24, 0.16 mM DMG and 8.2 ![]() oxine.
oxine.
Experimental conditions: see above table.
Experimental procedure
1. Demonstration of the use of mixture of ligands
a) Blank solution containing DMG only
as ligand
Add to the cell 5 ml of deionized water, 1 ml of 0.5 M ammonia buffer, 0.1 ml of 0.01 M DMG solution. Record a stripping voltammogram of the background according to the conditions for adsorptive stripping voltammetry, summarized in the table above.
b) Adding Ni2+ and Co2+
to the blank
Prepare by stepwise dilution 5 ml of diluted separate
solutions of Ni2+ and Co2+ from the respective 1.7 mM
(100 ppm) stock solutions: 1 ppm (17 ![]() ) in 10 mM HCl, 0.2 ppm (3.4
) in 10 mM HCl, 0.2 ppm (3.4 ![]() ) in 10 mM HCl, and 0.02 ppm (0.34
) in 10 mM HCl, and 0.02 ppm (0.34 ![]() ) in 10 mM HCl.
) in 10 mM HCl.
Add 100 ![]() of each 0.02 ppm
solution to the cell containing the blank. Run a stripping voltammogram. Well
defined peaks of Ni and Co should appear.
of each 0.02 ppm
solution to the cell containing the blank. Run a stripping voltammogram. Well
defined peaks of Ni and Co should appear.
c) Adding Cu2+, Pb2+,
Cd2+ and Zn2+
Following section 1b, add to the cell 100 ![]() of a freshly prepared
solution containing 1
of a freshly prepared
solution containing 1![]() of each of Cu2+,
Pb2+, Cd2+ and Zn2+ in 10 mM HCl. Record a
stripping voltammogram. Do you observe any change as result of the presence of
the added metals?
of each of Cu2+,
Pb2+, Cd2+ and Zn2+ in 10 mM HCl. Record a
stripping voltammogram. Do you observe any change as result of the presence of
the added metals?
d) Adding oxine
Add to the above cell 50 ![]() of 1 mM oxine
solution. Record a stripping voltammogram. Note that the peaks of all the
species appear only in the solution containing the mixture of ligands.
of 1 mM oxine
solution. Record a stripping voltammogram. Note that the peaks of all the
species appear only in the solution containing the mixture of ligands.
2. Test for linearity of analytical signal vs. accumulation time
It is important to verify the range of accumulation time over which linearity is achieved. Perform the test for linearity for the metal to be analyzed in the following experiments.
Rinse the cell. Add to the cell 5 ml of deionized water, 1 ml of 0.5 M ammonia buffer, 0.1 ml of 0.01 M DMG. Record a stripping voltammogram of the background.
Add to the cell 100 ![]() of 0.02 ppm Ni2+
prepared in the previous experiment. Run a series of voltammograms for
different values of the accumulation time over the range of 20 to
80 sec.
of 0.02 ppm Ni2+
prepared in the previous experiment. Run a series of voltammograms for
different values of the accumulation time over the range of 20 to
80 sec.
Plot the analytical signal (sum of the slopes at the inflection points) vs. accumulation time.
3. Test for linearity of analytical signal vs. concentration (calibration curve)
In order to validate the use of the standard addition method, it must be shown that the peak current of the stripping voltammogram depends linearly on the concentration of the element. For this purpose run a series of voltammograms with increasing concentrations of the analyte, and plot the calibration line over the concentration range of 8 – 40 nM of the metal.
Add to the cell 5 ml of deionized water, 1 ml of 0.5 M ammonia buffer, 0.1 ml of 0.01 M DMG. Record a stripping voltammogram of the background for an accumulation time of 40 sec.
Add to the cell 100 ![]() of 0.02 ppm Ni2+
prepared in the previous experiment and run a voltammogram. Repeat this step with
four more additions.
of 0.02 ppm Ni2+
prepared in the previous experiment and run a voltammogram. Repeat this step with
four more additions.
Estimate the values of the analytical signal from the
voltammograms. Plot the analytical signal (corrected for background) vs.
concentration of the metals in the cell.
3.1.
Quantification of species
(a) The quantification is carried out by the standard addition method, which should be performed in the concentration range, where linearity of the calibration curve has been verified. In the analysis of samples with unknown concentration of analyte, the first step is to estimate the concentration level of the unknown by measuring the analytical signal and comparing it to the calibration curve constructed early.
_ If the signal is in the range of the calibration curve, proceed by the standard addition method.
_ If the signal of the unknown is larger, there are several possibilities:
(a) decrease the time of accumulation (not lower than 20 s);
(b) use a smaller volume of the sample for introduce into the cell;
(c) dilute the sample;
(d) check the linearity at higher concentrations by building a calibration curve in a higher concentration range.
_ If the signal is lower, the possibilities are:
(a) increase the time of accumulation;
(b) increase the volume of the sample introduced into the cell;
(c) if the detection limit permits, construct a calibration curve in a lower concentration range.
(b) Always perform first a measurement of the analytical signal in the background. For obtaining results with reasonable accuracy the signal of the unknown should be at least twice the value of the background.
4. Applications
4.1. Determination of contamination of water by Ni2+ as a result of contact with stainless steel
Stainless steel being in contact with hot water release traces of Ni. The contamination of Ni is higher in presence of acids.
This experiment can be performed with any stainless steel equipment: any kind of vessels, heating elements for preparing water for hot drinks, kettles and pots with stainless steel parts, etc.
To prepare the samples, heat 100-200 ml of deonized water containing 10 mM NaCl (the addition of the salt is to simulate drinking water). Leave the hot water in contact with the stainless steel for about 20-40 min. Use this solution to determine the amount of Ni2+ leached from the stainless steel.
Add to the cell 5 ml of deionized water, 1 ml of 0.5 M ammonia buffer, 0.1 ml of 0.01 M DMG. Record the stripping voltammogram of the background.
Add to the cell 0.05 – 2.0 ml of the analyte solution and record a voltammogram. The aliquot of the unknown is determined by the concentration of the analyte: the larger the concentration – the smaller the portion of added sample (cf., note 3.1.). Make at least two standard additions, using the proper diluted solution of Ni2+ (0.02 ppm or 0.2 ppm) and run the voltammograms again. Consult with the instructor about the volume of the standard additions.
Estimate the values of the analytical signal from the voltammograms. Plot the analytical signal corrected for background vs. concentration of the metal in the cell. Report the concentration of the metal in the sample in mol/l and ppb. What is the pH of the analyte sample?
4.2. Determination of contamination of water by Ni2+ as a result of contact with a nickel strip
Prepare a sample by inserting a Ni strip (total area about 60 cm2) into a 100 ml glass beaker filled with deionized water. Wait for about 20-30 min. It is possible to use hot water for shortening the waiting time. Use this solution to determine the amount of Ni2+ leached from the strip.
Add to the cell 5 ml of deionized water, 1 ml of 0.5 M ammonia buffer, 0.1 ml of 0.01 M DMG. Record the stripping voltammogram of the background.
Add to the cell 0.050-0.100 ml of the analyte solution and record a voltammogram. Make at least two standard additions, using the 0.2 ppm solution of Ni2+ and run the voltammograms again. Consult with the instructor about the volume of the standard additions.
Estimate the values of the analytical signal from the voltammograms. Plot the analytical signal corrected for background vs. concentration of the metal in the cell. Report the concentration of the metal in the sample in mol/l and ppm.
4.3. Direct determination of Ni2+ in drinking (tap) water
Add to the cell 5 ml of deionized water, 1 ml of 0.5 M
ammonia buffer, 0.1 ml of 0.01 M DMG. Record the stripping voltammogram of the
background.
Rinse the cell and run another voltammogram for the solution containing 3 ml tap water, 2 ml deionized water and the same amounts of ammonia buffer and DMG. Use the method of standard additions, as in the previous applications, for quantitative determination of Ni2+ in the sample of tap water. Report the concentration of the metal in tap water in mol/l and ppb. Compare your results with the official permissible levels.
4.4. Contamination of water by Pb2+
as a result of contact with glazed ceramic vessel
Lead compounds have been and are still used in the manufacture of ceramic glazes and crystal glasses. Certain compounds of lead, particularly brightly colored lead oxides, have been used for leaded glasses and leaded glazes on ceramics for thousands of years. The addition of lead improves the appearance and cutting properties of crystal glass. Small amounts of lead are also present in optical glass. The major application of leaded glass is in television screens and computer monitors, and serves to protect viewers from the harmful X-rays generated by these appliances. Lead-containing glazes are used in some pottery, tiles and tableware. On contact with water or other solutions, the lead is slowly released in trace amounts, especially in acidic solutions.
To read more about the
properties of lead Go
to
http://www.ldaint.org/factbook/factbookch1.htm
http://ianrpubs.unl.edu/water/g1333.htm
On a hot plate, heat about 100 ml of deionized water in a glass beaker until the water boils, add 1 ml of 10 mM HCl (pH 4). Transfer the hot water to a glazed ceramic vessel, wait for about 20 min. Determine the degree of contamination by Pb2+ in this solution.
Add to the cell 5 ml of deionized water, 1 ml of 0.5 M
ammonia buffer, 0.1 ml of 0.01 M DMG and 50 ![]() of 1 mM oxine
solution. Record the stripping voltammogram of the background.
of 1 mM oxine
solution. Record the stripping voltammogram of the background.
Add to the cell 0.1 – 0.2 ml of the analyte solution and
record a voltammogram. Make at least two standard additions, using the 1![]() solution of Pb2+ and
run the voltammograms again. Consult with the instructor about the volume of
the standard additions.
solution of Pb2+ and
run the voltammograms again. Consult with the instructor about the volume of
the standard additions.
Estimate the values of the analytical signal from the voltammograms. Plot the analytical signal corrected for background vs. concentration of the metal in the cell. Report the concentration of the metal in the sample in mol/l and ppm.
4.5. Contamination of water by Pb2+
as a result of contact with crystal glass
Crystal glasses contain up to 30 % lead oxide, PbO. The European definition of lead crystal as glass that contains a minimum of 24% lead oxide is a feature that once contributed to its reputation for quality. The purpose of lead oxide is to increase the density of glass, which in turn, increases the refractive properties. When it is cut and polished, it creates more colors and sparkle than regular glass.
On a hot plate, heat about 100 ml of deionized water in a glass beaker until the water boils, add 1 ml of 10 mM HCl (pH 4). Transfer the hot water to a crystal glass, wait for about 20 min. Use this solution as analyte for the determination of Pb2+.
Add to the cell 5 ml of deionized water, 1 ml of 0.5 M
ammonia buffer, 0.1 ml of 0.01 M DMG and 50 ![]() of 1 mM oxine
solution. Record the stripping voltammogram of the background.
of 1 mM oxine
solution. Record the stripping voltammogram of the background.
Add to the cell 0.1 – 0.2 ml of the analyte solution and
record a voltammogram. Make at least two standard additions, using the 1![]() solution of Pb2+ and
run the voltammograms again. Consult with the instructor about the volume of
the standard additions.
solution of Pb2+ and
run the voltammograms again. Consult with the instructor about the volume of
the standard additions.
Estimate the values of the analytical signal from the voltammograms. Plot the analytical signal corrected for background vs. concentration of the metals in the cell. Report the concentration of the metal in the sample in mol/l and ppm.
4.6. Determination of traces of Pb2+
released in water as a result of contact with brass parts used in
water-delivery lines
Brass is an alloy of copper, containing mainly copper and zinc. It is used in connecting parts of water-delivery lines. Small amounts of lead are sometimes added to improve the mechanical characteristics of the alloy.
Lead rarely occurs naturally in water. Most lead contamination takes place at some point in the water delivery system. This occurs as a result of corrosion, the reaction between the water and lead in parts of the water-delivery system. Components of water-delivery systems which may contain lead include service connections, pipes, solder and brass fixtures. Lead in drinking water from plumbing is most often a problem in either very old or very new houses. Many household faucets, plumbing fittings, check valves and well pumps are manufactured with brass parts. While brass contains some lead to make casting easier and the machining process more efficient, the lead content of brass plumbing components is now restricted to a maximum of 8 %. Even at this low level, however, lead can be leached from new brass faucets and fittings. Eventually, if the water is not corrosive, hard-water minerals deposit on the interior of the plumbing. These deposits form a calcium carbonate lining inside pipes and fittings, which protects against lead contamination. It may take up to five years for an effective calcium carbonate lining to form. Softening naturally hard water with an ion-exchange water softening unit can either prevent or dissolve the calcium carbonate scale, eliminating its possible protective effect.
To read more about the
properties of copper Go
to
http://ianrpubs.unl.edu/water/g1360.htm
Place in a beaker a brass connector from a water-delivery line and fill with deionized water at room temperature. Leave over night. Use this solution as analyte for the determination of traces of Pb2+.
Add to the cell 5 ml of deionized water, 1 ml of 0.5 M
ammonia buffer, 0.1 ml of 0.01 M DMG and 50 ![]() of 1 mM oxine
solution. Record the stripping voltammogram of the background.
of 1 mM oxine
solution. Record the stripping voltammogram of the background.
Add to the cell 0.1 – 0.2 ml of the analyte solution and
record a voltammogram. Make at least two standard additions, using the 1![]() solution of Pb2+ and
run the voltammograms again. Consult with the instructor about the volume of
the standard additions.
solution of Pb2+ and
run the voltammograms again. Consult with the instructor about the volume of
the standard additions.
Estimate the values of the analytical signal from the voltammograms. Plot the analytical signal corrected for background vs. concentration of the metals in the cell. Report the concentration of the metal in the sample in mol/l and ppm.
References
1. Carlo Colombo, Constant M. G. van
den Berg, Anal. Chim. Acta, 1997, 337, 29-40.
2. Miropi G. Paneli and Anastasios
Voulgaropoulos, Electroanalysis, 1993, 5, 355-373.
3. S. B. Adeloju, A. M. Bond and M. H. Briggs, Anal. Chim. Acta, 1984, 164, 181.
Go to Main Page