5. POLAROGRAPHY
5.1. Polarographic methods
In
direct current polarography (DCP) a constant potential is
applied during the entire drop-life time. A current-voltage curve is constructed
by applying a series of potential steps, each step being synchronized with the
drop fall. In most instruments, however, linearly changing potential is
applied, with a rate slow enough that the change of potential throughout the
drop-life time is about a few millivolts. The current is measured at the end of
the drop life.
|
|
|
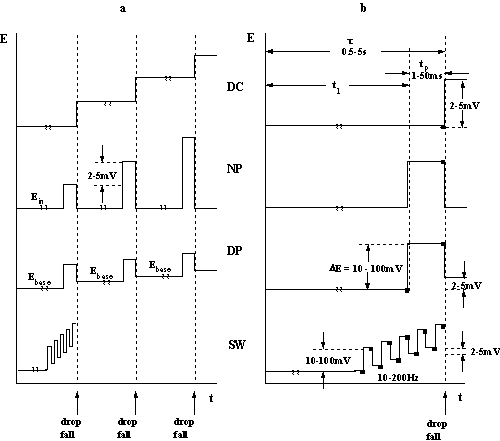
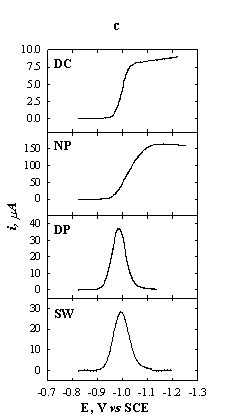
Fig.5-1 Schematic
presentation of some polarographic methods.
(a) Potential sequence of a polarogram.
(b) Potential sequence on a single drop (n current
sampling).
(c) Current-potential curves for 1 mM Zn2+ in 1 M KNO3.
DC: t = 2 s; NP: t = 2 s, tp = 5 ms; DP: t = 2 s, tp = 5 ms; DEp = 20 mV;
SW: delay time = 4 s, DEp = 20 mV, f = 100 Hz.
In
normal pulse polarography (NPP) the mercury-drop electrode is held for
most of its duration at a constant potential Ein, at which no
electrochemical reaction takes place under given experimental conditions. The
potential of interest Ep is applied in the last stage of the drop
life, for a length of time tp (of the order of few milliseconds).
The values of Ein and tp are kept constant throughout the
recording of the polarogram and Ep is changed from drop to drop
(Fig.5-1a).
The
limiting current in NPP is diffusion controlled. The experimental requirements
for diffusion control are the same as those for DC polarography. Since tp
is of the order of milliseconds, the diffusion layer thickness is very small
compared to the radius of the mercury drop reached at the end of its life. The
equations for planar diffusion can be applied with much better agreement than
for DC polarography (for tp = 1 ms, the diffusion layer thickness is
about 2·10-4 cm, while the radius of the
mercury drop is about 0.05 cm at t = 5 sec and m = 1 mg/sec). Furthermore, the area of the drop is
virtually constant during the application of the pulse (t is much larger than tp). The
constancy of the area implies also that no correction factor for the expansion
of the diffusion layer is required (the (7/3)1/2 factor). Thus,
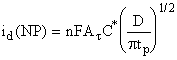 (1)
(1)
For comparison,
the current for DC polarography at time t is
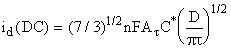 (2)
(2)
The current under
NP conditions is larger than that under DC conditions. The enhancement of the
current is
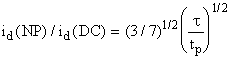 (3)
(3)
For typical
conditions, t = 5 s and tp = 5 ms, this ratio
is about 20. The contribution of the charging current is identical for both NPP
and DCP, and thus the detection limit of NPP is lower than that of DCP by an
order of magnitude.
The
i/E relationship for Nernstian processes at DCP and NPP is given by
![]() (4)
(4)
The detection
limit of NPP is about 2·10-7 M.
Differential
pulse polarography (DPP).
From analytical point of view, the sensitivity of DPP is even better than that
of NPP. The potential sequence on a single mercury drop and the potential
sequence used for recording an entire differential pulse polarogram are given
in Fig.5-1. The current is sampled twice during a drop life-time: (i) at t1,
just before the pulse, and (ii) at t, just before the drop fall. The polarogram
represents the current difference ![]() as function of
the base potential Eb. The curve is peaked shaped. The height of the
peak is proportional to the concentration of the electroactive species.
as function of
the base potential Eb. The curve is peaked shaped. The height of the
peak is proportional to the concentration of the electroactive species.
For
Nernstian processes Ox + ne = Red and C*red = 0, the faradaic
component of the current at the peak ![]() and Epeak are
and Epeak are
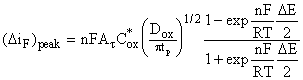 (5)
(5)
![]() (6)
(6)
The
peak form of the DP voltammogram is explained below. At sufficiently positive
potentials, Eb - E0 > (120/n) mV, the faradaic
currents ![]() and i(t1)
are both zero, and thus also
and i(t1)
are both zero, and thus also ![]() is zero. At
sufficiently negative base potentials, Eb - E0 <
(120/n) mV, the faradaic process proceeds at maximum rate and the
diffusion-limited current is reached; the current is independent of potential
and
is zero. At
sufficiently negative base potentials, Eb - E0 <
(120/n) mV, the faradaic process proceeds at maximum rate and the
diffusion-limited current is reached; the current is independent of potential
and ![]() s zero. At
potentials in the vicinity of
s zero. At
potentials in the vicinity of ![]() the current
difference
the current
difference ![]() is determined by
is determined by
![]() and has a finite
value.
and has a finite
value.
For
non-Nernstian processes ![]() is smaller than
for Nernstian ones. The sensitivity and the detection limit of DPP are better
for Nernstian processes, while for DC and NP polarography, they are independent
of the type of the processes. The reason for this can be understood by noting
that the equations for the limiting current in DC and NP polarography (eqs.1
and 2) do not contain assumptions about Nernstian processes. For the case of
DPP, however, the value of
is smaller than
for Nernstian ones. The sensitivity and the detection limit of DPP are better
for Nernstian processes, while for DC and NP polarography, they are independent
of the type of the processes. The reason for this can be understood by noting
that the equations for the limiting current in DC and NP polarography (eqs.1
and 2) do not contain assumptions about Nernstian processes. For the case of
DPP, however, the value of ![]() is derived from
the variation of the current around
is derived from
the variation of the current around ![]() The derivative
di/dE for non-Nernstian processes is smaller than for Nernstian, and results in
a lower
The derivative
di/dE for non-Nernstian processes is smaller than for Nernstian, and results in
a lower ![]() peak.
peak.
Although
(DiF)peak is smaller
than the limiting current at NPP, the detection limit at DPP is lower (about 10-7
M) due to the efficient compensation for the charging current.
Square
wave polarography (SWP). High sensitivity and low detection limit are
obtained with this technique. It is similar to DPP, however, the entire
potential sequence is applied during the life-time of a single drop. The
voltammogram is obtained in a few seconds, compared to much longer times with
the other techniques.
The
potential sequence is applied several seconds after the drop birth, in order to
take advantage of a larger surface area of the mercury drop.
As
in the case of DPP, the difference of the current before and after the
application of the pulse is measured. The performance of the method is better
for Nernstian processes than for non-Nernstian ones. Detection limit of the method
is about 10-7 M.
5.2. Handling the dropping-mercury electrode
(DME)
The
mercury capillary may be easily clogged and care must be exercised to prevent
contamination. The DME must never be allowed to stand in solution when mercury
is not flowing. Before allowing any solution to come in contact with the DME,
raise the leveling bulb and check that mercury drops are formed at the end of
the capillary. Allow the mercury flow during the entire laboratory session.
After use, the capillary should be washed thoroughly with distilled water. The
mercury reservoir is then lowered to stop the flow.
Note: BE AWARE! Mercury vapors are poisonous!
Notify the instructor in the event of mercury spill. Mercury should be
cleaned up immediately. Do not throw it down the drain.
Reference
electrode
An Ag/AgCl/1 M KCl
reference electrode is used. Its potential is -19 mV vs SCE.
Chemicals 1. 1 M KNO3
2. 0.1 M KNO3
3. 5.00 mM ZnCl2
4. 0.2 % Triton X-100
5. 0.01 M TlNO3
6. 0.01 M KIO3
7. 0.1 M NaCl
8. 0.1 mM Tl+, 0.1 mM Cd2+,
0.1 mM Zn2+, 0.1 mM Ni2+, 0.002 % Triton X-100
in 0.1 M NH4OH - 0.1 M NH4Cl
buffer
9. 0.5 M acetate buffer (pH 4.6)
10. 0.2 % ascorbic acid (freshly prepared)
Exp. 1. Oxygen waves
Fill the polarographic cell with 0.1 M KNO3
without passing the deaeration step. (Ask the instructor for help). Record the
polarogram using a voltage range of 0.2 to –1.8 V and a suitable current
sensitivity (![]() , tp = 20 ms, pulse amplitude = 20 mV, scan rate =
10 mV/s). Mark on the polarogram, what are the electrode processes that take place
at each wave. Estimate the concentration of dissolved oxygen, assuming that the
mercury flow rate is 1 mg/sec and D = 2·10-5
cm2/s.
, tp = 20 ms, pulse amplitude = 20 mV, scan rate =
10 mV/s). Mark on the polarogram, what are the electrode processes that take place
at each wave. Estimate the concentration of dissolved oxygen, assuming that the
mercury flow rate is 1 mg/sec and D = 2·10-5
cm2/s.
After
the first polarogram has been obtained, empty the cell, rinse and fill it with
a deoxygenated solution of 0.1 M KNO3. Run a polarogram of the
deaerated solution.
Hereafter,
all solutions for analysis should be deaerated.
Exp. 2. Polarographic spectrum
Introduce
into the polarographic cell the deoxygenated solution containing 0.1 mM of Tl+,
Cd2+, Ni2+ and Zn2+. Record the polarogram in
the potential range from -0.2 to -1.6 V, using several polarographic modes: DC,
NP, DP and SW. Compare E1/2 values with those found in the
literature.
Exp. 3. Capacitance currents
The
purposes of the experiments are:
(a) to observe the potential of zero
charge, EZ, at a dropping-mercury electrode by recording i vs
E curves in solutions containing supporting electrolyte only;
(b) to measure currents due to charging of
the double layer.
Record
a polarogram of a deaerated 0.1 M KNO3 solution at potentials ±200
mV around EZ (EZ = -0.45 V vs SCE, in absence of
specific adsorption). Use an expanded potential scale.
Record
a current-time plot for a single drop at E = EZ. Repeat for a
potential positive and a potential negative to EZ. Do not forget to
mark the zero current. Determine the value of EZ and compare with
data in literature.
Analyze
the current/time plots and compare with theory. Estimate from the current/time
plots the double layer capacitance, assuming m = 1 mg/sec.
Exp. 4. Maxima and maxima suppression
Run
a polarogram of 1 mM Zn2+ in 0.1 M NaCl. Repeat the experiment with
the same solution, containing 0.002% Triton X-100. Note the effect of the
detergent on both the peak and the limiting current.
Exp. 5. Migration currents
The
purpose of the experiment is to test the effect of the concentration of the
supporting electrolyte on the limiting current. For this purpose run
polarograms of 1 mM Tl+ solution with increasing concentrations of
KNO3 (0, 1, 2, 5, 10, 50, 100 mM). Use the potential range of –0.1
to 1.0 V (![]() , tp = 20 ms, pulse amplitude = 20 mV, scan rate =
10 mV/s). Plot the limiting current vs concentration of supporting
electrolyte. Compare with theory, assuming equal equivalent conductances for
all ionic species of interest.
, tp = 20 ms, pulse amplitude = 20 mV, scan rate =
10 mV/s). Plot the limiting current vs concentration of supporting
electrolyte. Compare with theory, assuming equal equivalent conductances for
all ionic species of interest.
Repeat
the experiment by replacing the Tl+ solution with KIO3.
In that case 0.002% Triton X-100 is required to suppress the polarographic
maxima. Use the potential range of –0.8 to -1.8 V. The electrode reaction of the
reduction of KIO3 is:
![]()
Exp. 6. Analysis of the polarographic wave
The
purposes of the experiment are:
(a) to verify that the limiting current of
the TI+ - reduction polarographic wave is diffusion controlled;
(b) to determine the half-wave potential
and the number of electrons involved in the reduction.
Prepare
0.5 mM solution of TlNO3 in 0.1 M KNO3. (The Tl+
concentration has been chosen low enough in order to neglect potential drop in
the solution). Run a polarogram in order to choose a potential in the limiting
current region. Record a current/time plot at the chosen potential. Prove, that
the current is totally diffusion controlled, and calculate the diffusion
coefficient, assuming that m = 1 mg/sec.
In
order to enable an accurate measurement of the currents along the wave, run an
additional polarogram at a slow scan rate (5 mV/s).
Determine
E1/2 of the Tl+ reduction wave and compare with
literature.
Determine
the number of electrons involved in the reduction process from E vs
log[(id -i)/i] plot.
Exp.
7. Quantitative determination of
Zn2+ in drinking water
Fill the cell with the sample of drinking
water and record a polarogram. You may prefer to use one of the more sensitive
polarographic methods (normal pulse, differential pulse or square wave). Make a
rough estimation of the concentration of Zn2+.
For
the analytical determination use both the standard addition method and the
calibration curve method. Show the instructor your detailed plan of operation,
including the composition of the solutions. Prepare the standard solutions in
50 ml volumetric flasks. For the calibration curve method use as the supporting
electrolyte the solution of 10 mM NaCl. Compare the results of the two methods
of determination.
Exp. 8. Determination of ascorbic acid (Vitamin C) in citrus juice by the standard-addition method.
Ascorbic
acid yields a well-defined polarographic oxidation wave. The determination can
be carried out directly in freshly prepared diluted juice as well as in
conserved citrus juice. The method of
standard additions is used for the quantitative determination of the
concentration of the Vitamin C.
|
|
|
|
Ascorbic acid |
Dehydroascorbic acid |
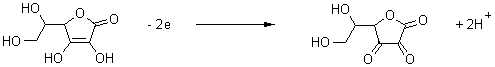
Procedure
1.
Calibration
curve
Prepare
a fresh stock solution of 50 ml 0.2 % ascorbic acid.
Prepare
five standard solutions of ascorbic acid in volumetric flasks of 25 ml. To each
one add 0.5 ml 0.5 M acetate buffer and different volumes of 0.2 % ascorbic
acid: 0, 200, 400, 600 and 800 ![]() . Dilute to the mark with distilled water.
. Dilute to the mark with distilled water.
For each
solution record a NP (![]() , tp = 20 ms, pulse amplitude = 20 mV, scan rate =
10 mV/s) and SW (
, tp = 20 ms, pulse amplitude = 20 mV, scan rate =
10 mV/s) and SW (![]() , tp = 20 ms, pulse amplitude = 20 mV, scan rate =
100 mV/s) polarograms over the potential range: -150 to +200 mV vs. Ag/AgCl/1 M
KCl, with Ein = -150 mV. Consult with the instructor about the
preferable mode of polarography to be used in further experiments.
, tp = 20 ms, pulse amplitude = 20 mV, scan rate =
100 mV/s) polarograms over the potential range: -150 to +200 mV vs. Ag/AgCl/1 M
KCl, with Ein = -150 mV. Consult with the instructor about the
preferable mode of polarography to be used in further experiments.
Plot
id vs. concentration of ascorbic acid. Is the plot linear and does
it pass through the origin? On the basis of these observations decide if the
standard-addition method is applicable.
2.
Determination
of ascorbic acid in citrus fruits
Squeeze an orange, grapefruit or lemon until about 10 ml of juice is obtained. Filter the juice through a porous funnel (pore size about 1 mm).
Prepare
four 25 ml volumetric flasks. Add to each, 0.5 ml of 0.5 M acetate buffer, 2.0
ml of the juice and standard additions of 0, 200, 400 and 600 ![]() of 0.2 % ascorbic acid. Dilute to the
mark with distilled water.
of 0.2 % ascorbic acid. Dilute to the
mark with distilled water.
Record polarograms under the same conditions
as in the calibration step.
Draw the standard additions plot and determine the
concentration of the analyte. Report the concentration of ascorbic acid
(Vitamin C) in the original sample (juice) in mol/l and ppm.
3.
Determination
of ascorbic acid in conserved citrus juice
Plan an experiment for the determination of ascorbic acid in preserved (commercial) citrus juice. Use the scheme of the previous experiment. Consult with the instructor as to the differences in procedure for preserved as opposed to natural juice.
Recommended
Literature
1. H.
H. Willard, L. L. Merritt, J. A. Dean and F. A. Settle, Instrumental Methods
of Analysis.
2. D. A. Skoog and
D. M. West, Principles of Instrumental Analysis.
3. D. A. Skoog and
J. J. Leary, Instrumental Analysis.
4. D. C. Harris, Quantitative
Chemical Analysis.
5. A. J. Bard and L.
R. Faulkner, Electroanalytical Chemistry.
Go to Main Page