1. TITRIMETRIC METHODS OF ANALYSIS
Titrimetric analysis consists in determining the number of moles of reagent (titrant), required to react quantitatively with the substance being determined. The titrant can be added (a) volumetrically, with a glass or automatic burette or with a low flow-rate pump, or (b) coulometrically, with an electrochemical generation from a proper electrolyte.
Titrimetric
methods of analysis have the virtue of being like gravimetric methods, absolute
in that the concentration of the substance in question is determined from the
basic principles of chemistry, and no calibration curves are required.
Various methods are available for end-point
determination: spectrophotometry, potentiometry, amperometry, conductometry,
etc. The potentiometric end-point determination is the most widely used.
1.1. Modes of titration
Titrations are performed manually
point by point, or automatically, where the titrant is
introduced continuously (monotonically or dynamically). In modern analytical
chemistry automation is of increasing importance. Automation of a
point-by-point titration is seldom trivial. Kinetic factors concerning the
chemical reaction and the response of the indicating system are of paramount
importance. Cell configuration, stirring, positioning of end-point detector and
of input of titrant are to be considered for ensuring high accuracy.
Equilibrium throughout the titration curve
can be attained only when the titrant is added at an infinitely low rate. Two
factors determine the rate at which a potentiometric curve approaches the state
of equilibrium: the kinetics of the chemical reaction and the kinetics of the
electrode process. Both factors are slowest in the vicinity of the equivalent
point (why?), the region with utmost importance, from which the end point is
determined (Fig.1-1a). (In conductometric titrations, on the contrary, the
region around the end point is of no analytical importance; the end point is
determined by extrapolation from the left and right branches of the titration
curve away of the end point). At faster titration rates compared to the
kinetics of the system, a delayed end point is obtained (Fig.1-1b).

Fig.1-1 Continuous potentiometric titrations
The degree of the delay of the end point
depends on the concentration of analyte, the composition of the solution, the
concentration of titrant and the rate of the titrant adding. The larger the
concentration of the analyte, the smaller the delay. The larger the amount of
titrant added per unit of time, the larger the delay.
Several strategies are used to virtually
eliminate the delay:
(a) Modifying the rate of titrant adding,
either by reducing it throughout the entire titration curve - the
monotonic mode, or by reducing it only along the vicinity of the
equivalence point - the dynamic mode (Fig.1-1b; the delay of the
dynamic curve is exaggerated for the purpose of the illustration). With the
dynamic mode, the rate of the titration is inversely proportional to the rate
of potential change, ![]() (Fig.1-1c),
where l is the progress of the titration.
(Fig.1-1c),
where l is the progress of the titration.
(b) Performing a repetitive-monotonic
titration1 (Fig.1-1d). In this mode a series of titrations
is performed in a consecutive manner. The measurements are made under
non-equilibrium conditions. The titrant is introdused, or electrochemically
generated, at a fast and constant rate. Each titration is discontinued at some
time after the end point (the actual time is of no importance). A new portion
of sample is added to the cell and the titration is resumed at the previous
rate. The new aliquot can be added also without suspending the flow of the
titrant. The end points of the titrations are usually delayed. Each end point in
a series of titrations is delayed only once, by a period of time depending on
the specific type of titration. Previous delays are not accumulated. The delay
of the previous titration is always nullified a short time after a new portion
of an analyte is added, since the response of the system is slow only around
the equivalence point, but not far away from it.
The
basic requirement for a successful series of consecutive titrations is the
reproducibility in the delay of the response around the end point of the
individual titrations. When the delay remains constant, the difference between
two successive equivalence points is the same as the difference between two
successive end points. In such case the measurable difference between the nth
and (n-1)th end point leads to the concentration of the nth
sample.
The
repetitive-monotonic mode plays a double role: (i) canceling delay due to slow
(but reproducible) electrode response, (ii) fulfilling the role of a
pretitration, in which traces of chemically active impurities in the background
are neutralized.
All
modes of automatic titrations have to be tested for accuracy if no previous
knowledge is available (what test would you suggest for the
repetitive-monotonic titration?).
1.2. Pretitration
In
trace analysis, it is common practice to neutralize chemically active species
present in the background prior to the titration of the analyte. The principle
of pretitration is as follows: a small excess of titrant is introduced into the
background solution, and the signal of the indicating system is measured. Then
the analyte is added and titrated till the intensity of the signal of the
indicating system equals to that of the pretitration step. This simple form of
pretitration has been used in the determination of As(III) with
electrogenerated I2. A small excess of iodine is
electrogenerated till a light blue coloration with the starch indicator is
formed. The sample is then introduced and titrated till the same intensity of
color is reached. The pretitration serves here two purposes: one is to oxidize
possible impurities in the buffer, and the other is to take care of the fact
that in very dilute solutions (with respect to the analyte) the change of color
of the indicator is not abrupt, but gradual. A suitable transition,
recognizable by the human eye, is chosen as end point.
1.3. Manual volumetric titrations
The classical method of titration comprises
in manual introdusing of titrant using glass burettes or piston burettes.
It
is recommended to run a fast preliminary titration in order to estimate the
location of the end point and the magnitude of the potential jump. The first
few additions of titrant may be rather large, and when the potential begins to
change rapidly, the size of the additions should be decreased. In the
neighborhood of the equivalence point the additions should be reduced to (Ve.p./500)
ml and time should be allowed for stable reading to be achieved. In following
titrations the titrant may be added in large amounts almost up to the equivalence
point. If an automatic burette is used, equal additions around the end point
are recommended.
1.4. Automatic volumetric titrations
In modern analytical practice automation of
titrations is essential for the following reasons: (i) convenience and speed,
(ii) performing analysis without supervision, (iii) enable application of
elaborate techniques for analyzing the data (with computerized systems).
Several
devices for automatic transfer of titrant can be used:
·
Piston
burettes are highly
reliable, do not require calibration, but are expensive.
·
Peristaltic
pumps are of relatively
low cost and highly versatile. However, they require frequent calibration due
to the continual changes in the physical properties of the flexible tubes
employed (ID of the tubes is about 0.5 mm). It is recommended that the
calibration be performed at the same time as the titration.
Note: Flexible tubing is the only
sensitive element in the peristaltic pump. Potential leaching of plastisizers
or bland materials is to be taken into account when working with
polypropylene-based tubing (PharMed or Norprene). Viton tubing withstands harsh
solvents and aggressive chemicals, but has a relatively short lifetime.
·
Suction-stroke
piston pumps (metering pumps) are as versatile as the peristaltic pumps, but more precise and require
less calibration.
The peristaltic and the suction-stroke pumps have several common advantages: simplicity of operation, simple control of flow rate, suitability for automation, convenience in exchange of titrant, low wastage of reagents. However, there are limitations in the use of corrosive reagents.
All
these devices enable work in a wide range of flow rates of titrant transfer.
For titrations in small volumes low flow rates are used (0.1 - 1 ml/min).
An
experimental setup for potentiometric titration using a peristaltic pump is
shown on Fig.1-2.
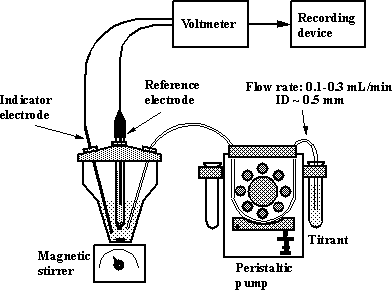
Fig.1-2 Experimental setup for a potentiometric titration using a peristaltic
pump as titrator.
1.5. Location of end point
There are several methods of end-point
determination. We shall mention only few of them.
For
potentiometric indicating system with a reference electrode (Fig.1-3) the end
point is determined from the first derivative of the titration curve. For the indicating
system with two identical electrodes, the end point is determined directly from
the titration curve.
It
is advised to attempt to estimate the end point with a precision of 0.2% or
better.
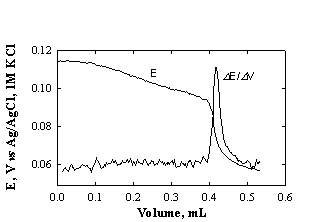
Fig.1-3 Titration and derivative curves for determination of Ca2+ with EDTA.
Sample: 1 ml drinking water;
titrant: 5 mM EDTA; pH 10.5; total volume ~5 ml; indicating electrode:
mercury-coated silver electrode, reference electrode: Ag/AgCl/1 M KCl.
(Data from Exp.1 in section
"Potentiometric titrations").
Repetitive-monotonic mode of titration
In continuous follow-up of the progress of a titration, the main problem is the slow response of the electrodes, mainly in the vicinity of the end point. In commercial instrumentation the rate of titration is decreased when approaching the end point. A different (simpler) approach is proposed here.
The
titration is carried out at constant rate (constant generating current).
Due to the slow response of the electrode, the end point lags behind the
equivalence point. Performing a series of titrations by adding the sample to
the previously titrated solution solves the problem (Fig.1-4).
The distance between the successive end points corresponds to the
true value of the end-point time. (This is correct only for cases in which the
response of the electrodes does not change from titration to titration).

Fig.1-4 Repetitive-monotonic coulometric titration of chlorides.
Initial composition: 0.500 ml
5.10 mM NaCl, 0.25 ml 1.2 M HClO4, 4 ml methanol.
Generating electrodes: a
mercury-coated silver wire and a small-area tungsten electrode (0.1 cm2);
ig = 2 mA.
Potentiometric indicating
system: twin mercury-coated silver wires, iind = 2 mA.
End-point times are printed;
first end point is delayed. Arrows correspond to new additions of analyte.
Treatment of potentiometric titration curves for the case of incomplete reactions at the equivalence point. Gran's approach
The end point in a potentiometric titration
is taken as the inflection point of the titration curve. At low concentration
of analyte or insufficiently large equilibrium constant, the degree of
incompleteness of the reaction is highest around the equivalence point (why?).
Gran's approach is based on transformation of the potentiometric plot (in which the signal is a logarithmic function of the concentration) to a plot where the transformed function is linear in concentration. Extrapolation of the linear part enables a more accurate determination of the end point. Different types of transformation are available in the literature.
Example of Gran's treatment is presented in
Fig.1-5. Vt/Vin is a correction for the dilution. (Vt
is the volume of the titrated solution in the cell at a given time t, Vin is the initial volume in the cell prior
to the start of the titration).

Fig.1-5 Determination of bicarbonates in drinking water.
Sample: 5 ml tap water from
Tel-Aviv University. Titrant: 10.00 mM HCl.
We shall apply
the Gran treatment to the potentiometric titration of a weak base (bicarbonate)
with strong acids.
![]() (1)
(1)
The
titration is conducted by measuring the pH with a glass electrode.
The
titration curve, expressed as pH or E vs volume of titrant, yields an
S-shape plot. The end point is determined from data around an ill-defined
inflection point (the later results from an incomplete reaction between the
weak base and the strong acid; cf., Fig.1-5, left). If however, the titration
curve is displayed as [H+] vs volume of titrant, it consists
of a pair of straight lines, with the end point at the intersection of the two
branches (Fig.1-5, right).
The
form of the [H+]/volume plot is explained as follows. Previous to
the end point, [H+] is virtually zero and the left branch of the
titration curve is a straight line with zero slope. In the vicinity of the end
point the plot is curved, due to the incompleteness of the reaction. Beyond the
end point the carbonic acid is totally undissociated and the concentration of H+
is proportional to the number of moles of H+ added, yielding a
rising straight line. The advantage of the right-side plot in Fig.1-5 is
obvious.
The
transformation from one type of titration display to another is achieved using
eq.2:
![]() (2)
(2)
where k’ is a
constant, s is equal to (2.3RT/F) and fH+ is the activity
coefficient.
From eq. (2): ![]() , where
, where ![]()
For
applying the treatment, k and s should be accurately known. These quantities
can be determined from a blank titration, performed under conditions similar to
those of the sample titration (temperature, ionic strength, flow rate of
titrant, etc.) and as near as possible to the time of the main titration.
1.6. Operation with low flow-rate pumps
Calibration:
determination of the flow rate. The exact amount of titrant used in a titration is calculated upon the
value of flow rate of the pump and the time of titration. The calibration
consists of determination of the flow rate of the pump and represents the
amount of liquid transferred through the flexible pump tube during a measured
period of time. It is recommended to perform the calibration as near as possible
to the time of the analysis. To achieve a statistically acceptable result, the
measurement must be repeated several times. The values of the mean and the
standard deviation (STD) of such group of measurements are the important
factors, which mainly determine the precision of the method. In the case of a
peristaltic pump the flow rate should be measured frequently due to changes in
physical properties of the flexible tubing.
The
arrangement used for calibration is shown in the figure below. Two 10-ml plastic
test tubes are located at both sides of the pump: one of them is filled with
deionized water, the other (the recipient vessel) is empty. A flexible pump
tube connects between the two test tubes. Note, that one end of the flexible
tube is dipped into the water, while the other end is placed into an additional
small Teflon tube and touches its inside wall. The purpose of the small Teflon
tube is to prevent loss of liquid while removing the pump tube at the end of
the measurement.
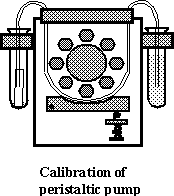
To perform the
calibration, place the recipient vessel (the empty 10-ml test tube with the
small Teflon tube inside) in a beaker and weigh both of them. Gently touch the
end of the pump tube with soft tissue to remove excess of liquid. Insert
carefully the end of the pump tube into the Teflon tube. Turn the pump on and
transfer deionized water for a predetermined time (~1 min). Stop the pump and
remove the pump tube from the recipient test tube. Weigh again. Calculate the
flow rate in g/min and ml/min. Repeat the calibration three more times.
Operating
parameters for peristaltic pump. The required flow rate of titrant is
achieved by choosing the proper tubing size and rotation rate of the
peristaltic pump. For optimal operation the pressure applied on the flexible
tube is chosen in the plateau region of the experimental plot in Fig.1-6. The
shape of the plot depends on size and type of tubing material.

Fig.1-6 Effect of pressure applied by the micrometer screw on the flexible tube
of a peristaltic pump.
Adding
the titrant. Remove any
drops on the outside of the pump tube with absorbent tissue and insert the tube
into the titrant. To fill the pump tube with the titrant turn the pump on and
let the titrant flow through the tube for about 30 s.
At
the end of the working session the pump tube should be rinsed with
distilled water. Rinse the free end of the tube with water. Dip it into
distilled water and operate the pump for several minutes. For the peristaltic
pumps after rinsing the tube release the tension on the tube. This prolongs its
life.
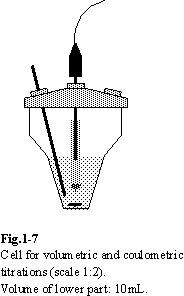
1.7. Cell
The geometry of the titrating cell is of
lesser importance in point-by-point titrations. However, for automatic mode of
titrations several requirements should be fulfilled. The stirring should be
efficient, but smooth. Vortexes and air bubbles should be avoided in order to
keep the relevant parts of the sensor in constant touch with the solutions. The
volume of the solution in the cell and the intensity of the stirring should be
such as to ensure efficient transport throughout the solution. The cell shown
in Fig.1-7 is found to be convenient for automatic titrations (in this specific
cell the volume of the solution should not extend beyond the lower part of the
cell).
Components inserted into the cell should not
touch the cell walls or each other. Such situation would cause formation of
pockets of stagnant solution, perturbing the distribution of reactants. In
volumetric titrations the tip of the burette (Teflon tube with inner diameter
not larger than 0.8 mm or antidiffusional microvalves) should be dipped into
the analyte solution, but not in the immediate vicinity of the indicator
electrodes. It is important to position it in the bottom part of the cell to
ensure fast mixing of the titrant. The tip, if not antidiffusional, should be
inserted just before the start of the titration.
Reference
1. D. Tzur and E. Kirowa-Eisner, Anal. Chim. Acta, 355,
85(1997).
Recommended Literature
1. D. A. Skoog, Principles
of Instrumental Analysis.
2. D. A. Skoog and
D. M. West, Principles of Instrumental Analysis.
3. D. A. Skoog and
J. J. Leary, Instrumental Analysis.
4. D. C. Harris, Quantitative
Chemical Analysis.
5. G. D. Christian, Analytical
Chemistry.
Go
to Main Page