 Return to the tutorial menu.
Return to the tutorial menu.
|
Immune reactions are divided into two broad categories: Humoral immunity
Cellular immunity
HISTOCOMPATIBILITY ANTIGENSMajor histocompatibility complex (MHC) or HLA complex is on chromosome 6Class I antigens: A, B, and C; these are present on all nucleated cells and are linked to beta-2 microglobulin; they are tested for and detected by serologic means; in practice the "C" antigens are unimportant; a number of alleles are present, and each person inherits one from each parent; thus, a person might be HLA typed as:
Class II antigens are in the D region (DR) and have a narrow distribution (mostly on mononuclear inflammatory cells); they are detected by mixed lymphocyte assay Why are HLA important?
HYPERSENSITIVITY REACTIONSType I hypersensitivityAnaphylaxis: Prior sensitization has resulted in an immune response initially mediated by CD4 lymphocytes (of the Th2 variety) that promote mast cell proliferation and plasma cell production of IgE. The IgE becomes bound to mast cells in places such as respiratory tract mucosa. Encountering the allergen again leads to mast cell degranulation with release of primary mediators (such as histamine, serotonin) which cause vasodilation, bronchoconstriction, etc. and release of secondary mediators (such as leukotrienes, prostaglandin) which lead to inflammatory cell infiltrates. The process of mast cell degranulation is diagrammed below:  There are two forms of anaphylaxis:
Type II HypersensitivityComplement dependent reactions: Antibody is directed against antigen on cells (such as circulating red blood cells) or extracellular materials (basement membrane). The resulting Ag-Ab complexes activate complement (via the classic pathway), leading to cell lysis or extracellular tissue damage. 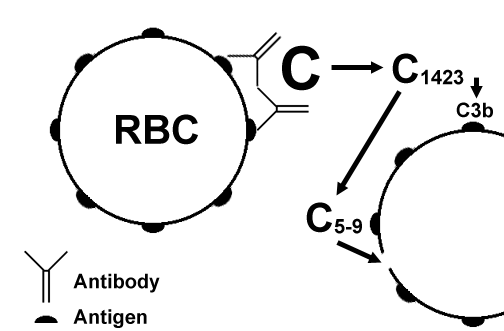 In the above diagram, a red blood cell has antigen fixed on its surface to which antibody attaches. The attached antibody sets off the complement cascade, which ends with the formation of the "membrane attack complex" of C5-9 which causes lysis of the cell. Other complement components may be generated, such as the opsonin C3b. Diseases in this complement dependent category include:
Antibody-dependent cell-mediated cytotoxicity (ADCC): Low concentrations of IgG or IgE (in the case of parasites) coat target cells. Inflammatory cells such as NK (natural killer) cells, monocytes, and granulocytes then bind to the immunoglobulin Fc receptors and lyse, but do not phagocytize, the target cells. 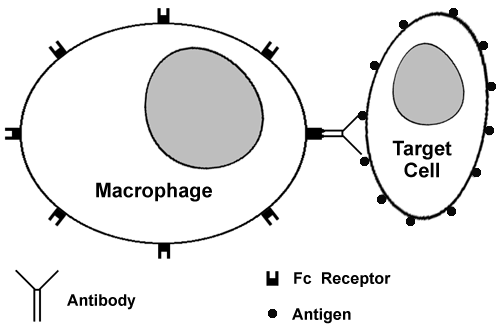 In the diagram above, a macrophage with Fc receptors on its surface is able to recognize a target cell coated with antibody via the Fc receptor portion of the attached antibody. The macrophage can then demolish the targeted cell by elaboration of proteases. Examples of ADCC include:
Antireceptor antibodies: IgG antibody is directed against receptors in target cells, resulting in complement-mediated destruction of the receptors. 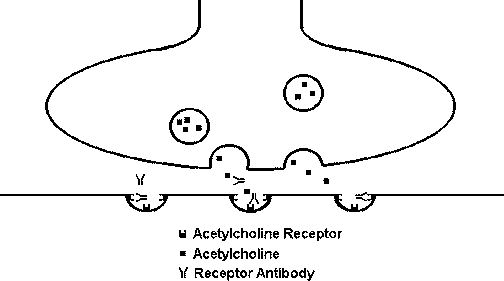 In the diagram above, antibody is directed against acetylcholine receptors at the motor end plate of a muscle, blocking the receptors and diminishing the muscular response. This is the mechanism for muscle weakness in myasthenia gravis. Diseases caused by this mechanism include:
Type III HypersensitivityThis reaction is mediated by immune (Ag-Ab) complexes which promote tissue damage primarily through complement activation (alternate pathway). C3b as an opsonin attracts neutrophils, which then release lysosomal enzymes. C5a as a chemoattractant brings in neutrophils. Serum complement is reduced as it is used up in this process. 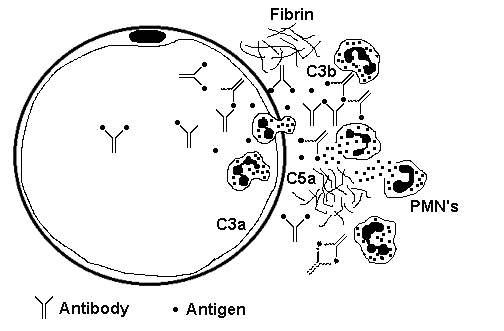 In the diagram above, antigen-antibody complexes are circulating and becoming trapped beneath the basement membrane of a small blood vessel, setting off the complement cascade and generating components that attract PMN's to generate an ongoing inflammatory response. Immune complexes can be deposited systemically or locally Systemic immune complex disease: Ag-Ab complexes form in the circulatory system and are deposited in tissues, typically near basement membranes in places such as blood vessels, glomeruli, skin, joints, pleura, and pericardium. Larger immune complexes are quickly phagocytized by macrophages and removed, but small to intermediate complexes formed with antigen excess may escape removal leading to:
Local immune complex disease: Also called an "Arthus" reaction, it occurs with local injection of the antigen and leads to focal vasculitis. This kind of immune reaction also plays a role in the development of hypersensitivity pneumonitis (so-called "farmer's lung"). Type IV HypersensitivityThis reaction is called "delayed hypersensitivity" because it is mediated by sensitized CD4+ T lymphocytes which process antigens in association with class II HLA molecules and release lymphokines. The lymphokines promote a reaction (especially mediated through macrophages) beginning in hours but reaching a peak in 2 to 3 days. 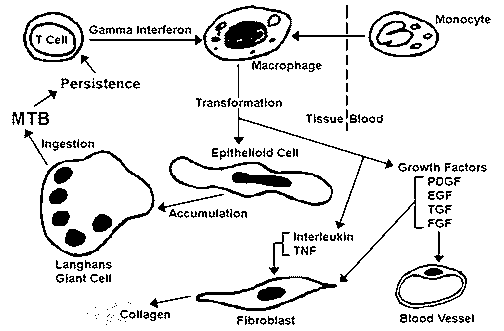 This diagram above illustrates a sequence of events in granuloma formation in response to Mycobacterium tuberculosis (MTB). The key cell in the process is the epithelioid macrophage. Hypersensitivity reactions with this mode of action include:
Cytotoxic T lymphocyte (CTL) mediated responses: CD8+ T cells are generated and lyse specific cells. Class I HLA molecules play a role. Reactions with this mode include:
TRANSPLANT REJECTIONImmunologic Mechanisms
Organ System Pathology
AUTOIMMUNE DISEASES"The fancier the plumbing, the easier it is to stop up the drain" -- The human immune system is very complex and, hence, there are numerous ways it can malfunction. There are a number of diseases that can result, and many of these have similarities. It is sometimes difficult to separate them. They are also often called "connective tissue" disorders because many of them are manifested in a variety of tissues. Hypersensitivity reactions involved in autoimmunity are primarily types II and III, though type IV reactions can also be present. Mechanisms proposed for development of autoimmunity include:Bypass of CD4+ T-cell tolerance of "self" antigens by:
Idiotype bypass through ligand mimicry, as seen in antireceptor antibody mediated disease, and cross-reactivity with infectious agents:
Antinuclear AntibodiesMany autoimmune diseases have serologic evidence for antibodies directed at components of "self" cell nuclei. Though these antibodies may not be the direct cause of, or evidence for, tissue injury, they are very useful for diagnosis and categorization of autoimmune diseases. The types include: ANA (antinuclear antibody): seen in many autoimmune diseases and not diagnostic of any. In general, the higher the titer, the worse the disease. There are some characteristic fluorescent staining patterns for ANA:
Though no autoantibody is completely sensitive or specific for a particular autoimmune disease, some of the strongest associations include:
Systemic Lupus Erythematosus (SLE)The manifestations are protean, and there is no characteristic or pathognomonic finding. Instead, this disease is diagnosed by finding suggestive serologic and clinical findings. Findings may include:
Genetic factors: tends to run in families; association with HLA Dr-2 and Dr-3; more common in young women (especially African-American). Drugs can produce "drug-induced" SLE: list includes procainamide, hydralazine, isoniazid, d-penicillamine. Discoid lupus erythematosus (DLE): a benign disease with skin involvement; ANA positive in only a third (but some of these go on to SLE). Mixed Connective Tissue Disease (MCTD)A wastebasket category for patients who do not clearly fit into other categories. There are features similar to SLE, scleroderma, and polymyositis. Most patients are middle aged females. Characteristic feature is a speckled ANA pattern. Scleroderma (Progressive Systemic Sclerosis, PSS)Characterized by excessive fibrosis in a variety of tissues from collagen deposition by activated fibroblasts. About 75% of cases are in women, mostly middle aged. Patterns of disease include: Limited scleroderm, or CREST syndrome: the benign form of PSS, serologically suggested by the presence of anti-centromere antibody
Diffuse scleroderma: the worst form of PSS; Scl-70 (anti-DNA topoisomerase I) antibody shows specificity for this form. It may include all of the findings with CREST, but additionally has renal findings: arterial intimal thickening and proliferation (hyperplastic arteriolosclerosis) leading to malignant hypertension with arterial fibrinoid necrosis, thrombosis, and renal infarction. Half of diffuse PSS patients die from renal disease. The lungs in this form of scleroderma may have diffuse alveolar fibrosis leading to honeycomb fibrosis Morphea: this is skin fibrosis only Polymyositis-DermatomyositisCharacterized by inflammation of skeletal muscle with weakness. Sometimes it is associated with a skin rash (hence, dermatomyositis). It is seen in ages 40-60, but also in ages 5-15, mostly in women. Some of these patients have Jo-1 antibody. Inflammation in polymyositis is mainly mediated by cytotoxic CD8 cells. In dermatomyositis, mainly antigen-antibody complexes produce a vasculitis in muscle and skin. Some adults (10-20%) develop cancer. Sjogren's SyndromeCharacterized by dry eyes and dry mouth as a result of lacrimal and salivary gland involvement by lymphocytic infiltration, fibrosis, and destruction mediated by CD4+ cells helping antibody production, of which anti-SS-A and anti-SS-B are the most specific. Most patients are middle to older age women. Lacrimal and salivary gland inflammation of any cause (including Sjogren's) is called Mikulicz's syndrome. PRIMARY IMMUNODEFICIENCY SYNDROMESX-linked Agammaglobulinemia of BrutonCongenital agammaglobulinemia (Bruton's Disease) results from a genetic defect on the long arm of X chromosome, so that males are primarily affected (inheritance occurs in an X-lined recessive pattern). The mutations affect production of a tyrosine kinase (Bruton tyrosine kinase, or btk) active in early pre-B cells which diminishes their maturation and leads to virtual absence of all immunoglobulin classes. Infants are observed to have multiple infections with bacterial organisms (Hemophilus, Staphylococcus), particularly in skin and lung. Agammaglobulinemia is the result of absent B-cells, but T-cell mediated immunity is intact. If affected persons survive, many will develop autoimmune diseases. Common Variable ImmunodeficiencyThis is a heterogenous group of disorders with an incidence of 1 per 100,000 that can involve both humoral and cell mediated immunity. Though there are normal numbers of circulating B lymphocytes, there is impaired secretion of one or more immunoglobulin isotypes, usually IgG or IgA. A selective abnormality of T cell activation, as demonstrated by decreased synthesis of interleukins (IL 2, 4, and 5) has been identified. Patients may have impaired gastrointestinal mucosal immunity. Another variant results from either a decrease in CD4 cells or an increase in CD8 cells. Also occuring is a variant resulting from the presence of T and B lymphocyte autoantibodies. At least two of the three main serum immunoglobulin isotypes are decreased. Persons with CVID are prone to recurrent bacterial infections, particularly sinusitis, bronchitis, pneumonia, bronchiectasis, and otitis. Bordatella pertussis infections occur in childhood. Viral infections are uncommon, though recurrent herpes simplex with eventual herpes zoster is an exception. Giardiasis is common. Half of CVID cases are diagnosed before age 21, but in some cases complications do not develop until adolescence or adulthood. There is an increased incidence of autoimmune diseases, particularly hemolytic anemia, thrombocytopenia, and pernicious anemia. In about two thirds of cases, normal numbers of circulating B lymphocytes are present. There is a decrease in immunoglobulins, generally in all classes, more often IgG and IgA, but sometimes only of IgG. DiGeorge SyndromeThe DiGeorge syndrome (or sequence) is a field defect of third and fourth pharyngeal pouch development in utero during organogenesis in the first trimester of pregnancy. A specific deletion on the long arm of chromosome 22 has been implicated. Anatomic structures that may be aplastic or hypoplastic include the thymus, parathyroid glands, great vessels, and esophagus. DiGeorge syndrome may be further subclassified as complete, in which there is almost total absence of thymic tissue, or as partial, in which there is only a decrease in thymic tissue Complete DiGeorge syndrome is characterized by normal levels of circulating immunoglobulin, though in some cases serum IgE is increased and IgA is decreased. However, affected children have markedly decreased numbers of circulating T lymphocytes, making them susceptible to fungal and viral infections. Children with partial DiGeorge syndrome have only a slight decrease in peripheral T lymphocytes and have increased infections, but with less frequency and with less severity than children with the complete form. Accompanying aplasia of parathyroid glands can lead to life-threatening hypocalcemia that may appear soon after birth. Severe Combined Immunodeficiency (SCID)SCID results from different defects with different inheritance patterns, but all demonstrate some degree of failure in development of both humoral and cell-mediated immunity. The major variants of SCID include:
There tends to be a greater decrease in cell mediated immunity than in humoral immunity. Normal or increased numbers of B lymphocytes may be present with the X-linked form, but these cells still do not function properly. There is very little serum IgG and virtually no IgM or IgA. Infants develop Candida skin rashes and thrush, persistent diarrhea, severe respiratory tract infections with Pneumocystis carinii (jirovecii) and Pseudomonas soon after birth, and failure to thrive after 3 months of age. Severe viral can occur. Maternal T lymphocytes crossing the placenta may produce graft versus host disease Wiskott-Aldrich SyndromeAn X-linked recessive pattern is seen because the defective gene is located on the short arm of the X chromosome (Xp11.23). The immunodeficiency is accompanied by thrombocytopenia and eczema. Circulating platelets are markedly decreased. T lymphocytes exhibit cytoskeletal disorganization and loss of microvilli by electron microscopy, and they express little CD43 by immunohistochemical staining. There is usually a normal level of serum IgG, along with a decrease in IgM, but often an increase in both IgA and IgE. The initial onset of disease in early childhood is accompanied by recurrent bacterial infections, particularly to encapsulated bacteria such as Streptococcus pneumoniae, with development of pneumonia, meningitis, and septicemia. Later, failure of T lymphocyte function may predispose to recurrent herpetic infections and to Pneumocystis carinii (jirovecii) pneumonia. A bleeding problem may result from the severe thrombocytopenia. Ataxia-TelangiectasiaA genetic defect is present on the long arm of chromosome 11 which predisposes to chromosome breakage and rearrangement, particularly on chromosomes 7 and 14, leading to a high risk for neoplasia and a marked sensitivity to radiation. This disorder is quite rare and has an autosomal recessive pattern of inheritance. There is a triad of progressive cerebellar ataxia, mucocutaneous telangiectasias, and recurrent respiratory tract infections with a variety of bacterial and fungal organisms. Immunoglobulin deficiencies, particularly IgA and/or IgE, may be present, though serum IgM is usually elevated. The symptoms usually begin between 9 months and 2 years of age. Selective IgA DeficiencyAbout 1 in 600 persons of European descent has a virtual lack of circulating IgA as well as secretory IgA which, in most cases, results from failure of the IgA type of B lymphocytes to transform into plasma cells capable of producing IgA or from impaired survival of IgA producing plasma cells. Some patients may have deficiencies in IgG subclasses 2 and/or 4, while IgG subclasses 1 and 3 are increased. Some patients go on to develop common variable immunodeficiency, suggesting that there is a similar defect in B cell maturation and function. Some affected patients are at increased risk for respiratory, gastrointestinal, and urinary tract infections, most often bacterial, and diarrhea is common. More severely affected persons may even have a sprue-like illness with malabsorbtion. Atopy, as demonstrated by asthma, can be present. Concomitant autoimmune diseases, particularly systemic lupus erythematosus and rheumatoid arthritis, can be present. About half of IgA deficient persons develop anti-IgA antibodies of the IgE type, so that transfusion of blood products containing serum with normal IgA levels leads to severe systemic anaphylaxis. Other Primary Immunodeficiency DisordersDeficiencies of other components of the immune system are uncommon. Some of the best known are:
|