- Case Presentation:
- History: A 9 month old infant boy has been seriously ill for the past month with severe respiratory difficulty. A tracheal aspirate grows Staphylococcus aureus. The baby also has diarrhea, and a stool culture grows Serratia marcescens. There is swelling in the right groin region from a large tender lymphadenopathy. A fine needle aspirate of the node shows numerous macrophages, and a culture grows Aspergillus fumigatus. Six months later there is a palpable lump in the right lower lateral neck region. The lump is excised and found to be an organizing abscess with granulomatous inflammation from which Staphylococcus aureus is grown. The baby was born at term to a G3 P3 23 year old woman whose prior two pregnancies resulted in normal term births. He appeared normal, with no congenital anomalies noted. However, the second child, a boy, died at at 2 from Burkholderia cepacia pneumonia. The first child is now a healthy 3 year old girl. A CBC on the infant shows:
-
| Hgb |
13.4 g/dL |
| Hct |
41.8% |
| MCV |
98 fL |
| Platelet ct |
277,000/microliter |
| WBC count |
8,120/microliter |
| WBC diff |
65 segs, 5 bands, 20 lymphs, 10 monos |
- Serum quantitative immunoglobulins show:
-
| IgA |
80 mg/dL |
(16 - 83 mg/dL) |
| IgG |
947 mg/dL |
(282 - 1026 mg/dL) |
| IgM |
101 mg/dL |
(39 - 142 mg/dL) |
- What do the findings suggest?
- There are normal leukocyte numbers and normal quantities of immunoglobulins. The pattern of infections is primarily bacterial. Aspergillus infections typically occur when there is a lack of neutrophils or neutrophil function. An inherited disorder of neutrophil function is suggested, such as chronic granulomatous disease.
- What tests could help confirm this suspicion?
- CGD is characterized by an absent or reduced function of the respiratory burst, which is the intracellular process in neutrophils that is dependent upon the enzyme NADPH oxidase, which produces oxygen free radicals used to kill phagocytized organisms. Flow cytometry can be employed to measure this respiratory burst. In the panel below at the left, unstained granulocytes display minimal measurable autoflourescence. When the laser-sensitive dye dihydrorhodamine123 is added, there is an increase in fluorescence from oxidation of the dye, as shown in the right panel:


- When phorbol myristate acetate (PMA) is added, normal granulocytes undergo an oxidative burst, oxidizing the dye and increasing the fluorescence, as seen in the left panel below. However, granulocytes from a patient with CGD do not display this burst, as shown in the right panel:
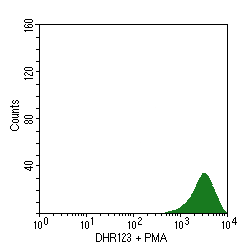
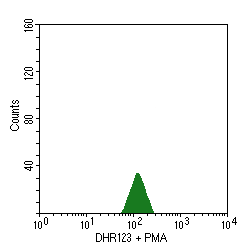
- The nitroblue tetrazolium (NBT) slide test is used for screening. In this test, patient neutrophils are exposed to a stimulus , incubated with NBT, and made into a smear on a slide. Under the microscope, the number of neutrophils with dark granules of reaction product are counted. Normally, more than 95% of the granulocytes will be positive, but in CGD <5% stain. The underlying defect is detected by more a more sensitive quantitative assay--the flow cytometry respiratory burst assay.
- Chronic Granulomatous Disease
- Explain the pathophysiology of this condition.
- There are five components of the NADPH oxidase enzyme. A defect in any one of these leads to diminished or absent enzyme function. Mutations occur in genes coding for proteins in the NADPH oxidative killing pathway for microorganisms ingested by granulocytes. Two-thirds of CGD cases are X-linked (the more severe form) and 1/3 autosomal recessive. There are normal numbers of circulating white blood cells. Most children present in infancy with recurrent bacterial and fungal infections. Lymphadenopathy is present with a granulomatous pattern of inflammation from recurrent infections with Aspergillus, Staphylococcus, Serratia, Nocardia braziliensis, and Pseudomonas, (Burkholderia). Organisms that make the catalase enzyme, such as S. aureus, are able to break down any hydrogen peroxide that forms. The granulomas form masses that can cause lumenal obstruction in the GI and urinary tracts.
- What can be done for affected children?
- Some children may be fairly healthy for long periods. Clean water and fresh food are essential to prevent infection, as is good dental care. However, any sign of infection must be promptly attended and cultures obtained. Aggressive antibiotic therapy is indicated. Use of gamma interferon therapy to increase neutrophil nitric oxide killing may be used in selected cases. Pain control for the various lesions and for the peripheral nerve inflammation is needed
- Case Presentation:
- History: A 2 year old child has had multiple episodes of pneumonia with Staphylococcus aureus and urinary tract infections with beta-hemolytic Streptococcus in the past several months. The child was born at term to a G1P1 woman. The baby appeared normal at birth and did well during infancy. However, the child has blond hair and blue eyes, while everyone else in the family has a darker complexion. The child bruises easily. A CBC on the child shows:
-
| Hgb |
13.7 g/dL |
| Hct |
41.3% |
| MCV |
84 fL |
| Platelet ct |
184,000/microliter |
| WBC count |
4,120/microliter |
| WBC diff |
45 segs, 2 bands, 39 lymphs, 14 monos |
- Examination of the peripheral blood smear shows giant dark granules in the neutrophils and lymphocytes.
- Peripheral blood smear showing features of Chediak-Higashi syndrome
- Serum quantitative immunoglobulins show:
-
| IgA |
69 mg/dL |
(16 - 83 mg/dL) |
| IgG |
823 mg/dL |
(282 - 1026 mg/dL) |
| IgM |
97 mg/dL |
(39 - 142 mg/dL) |
- What is suggested by these findings?
- There is a mild neutropenia with normal lymphocyte numbers and normal immunoglobulin levels. The history of bacterial infections and the giant granules suggests a disorder of neutrophils. The distincitive morphology is characteristic for Chediak-Higashi syndrome.
- Explain the pathophysiology of this condition.
- Mutation occurs in the LYST gene on chromosome 1q42 that encodes for a protein involved in intracecellular trafficking of proteins. Microtubules fail to form properly, and the neutrophils can't get their act together under chemotactic stimuli. There is abnormal fusion to form giant lysosomal granules that fail to function. Peripheral blood leukocytes have giant granules. Soft tissue abscesses with Staphylococcus aureus are common. Other cells affected by this disorder include platelets (bleeding), melanocytes (albinism), Schwann cells (neuropathy), NK and cytotoxic T cells (aggressive lymphoproliferative disorder).
- What is the clinical course of this condition?
- Recurrent infections are common. The infections can be treated with antibiotic therapy. The neuropathy gets worse with age and becomes disabling. Survival past the 20s and 30s is unlikely with the neuropathy. When the disease reaches the accelerated phase with aggressive lymphoproliferative disorder, death is likely. Bone marrow transplantation can be considered.
- Case Presentation:
- History: A baby is born at term to a G2P2 woman. The baby appears normal, and mother and baby are about to be discharged from the hospital, but the nurse in the newborn nursery notes that the baby's umbilical cord stump and the area around it on the abdomen is red and swollen. In fact, the umbilical remnant does not separate for nearly 2 weeks. The baby develops skin irritation where the diaper rubs against the skin, and this becomes ulcerated. Though no purulent exudate is seen, a culture of an area of skin necrosis grows Staphylococcus aureus. The baby develops a perirectal fissure a month later, and culture of this lesion grows Escherichia coli. A CBC shows:
-
| Hgb |
12.6 g/dL |
| Hct |
37.2% |
| MCV |
100 fL |
| Platelet ct |
222,000/microliter |
| WBC count |
98,660/microliter |
| WBC diff |
85 segs, 5 bands, 6 lymphs, 4 monos |
- Serum quantitative immunoglobulins show:
-
| IgA |
45 mg/dL |
(16 - 83 mg/dL) |
| IgG |
449 mg/dL |
(282 - 1026 mg/dL) |
| IgM |
71 mg/dL |
(39 - 142 mg/dL) |
- What do these findings suggest?
- The striking neutrophilia with the absence of a purulent exudate, despite presence of bacterial infection, is characteristic for leukocyte adhesion deficiency (LAD).
- What laboratory testing will confirm this suspicion?
- Flow cytometric analysis of neutrophils for CD11/CD18 will demonstrate CD18 deficiency.
- Explain the pathophysiology of this condition.
- Though neutrophils are present, even increased, in LAD, there is a problem with chemotaxis, so that neutrophils do not move to where they need to be. The problem is in neutrophil integrins, which are composed of alpha (CD11) and beta (CD18) subunits. Autosomal recessive mutations in CD18, the common beta chain of cellular integrins which aid in binding of leukocytes, are present. Leukocytosis is present, but there is absence of suppurative inflammation in areas of tissue necrosis and ulceration typically caused by Staphylococcus aureus, Candida, and gram negative enteric bacteria. Severe periodontitis with tooth decay is common in this disease.
- What is the clinical course of this condition?
- Recurrent infections are common and can be treated with antibiotic therapy. Most affected children die by the age of 5 if they do not receive a bone marrow transplant.
- Case Presentation:
- History: A 35-year-old man has been relatively healthy for most of his life, though he has had more upper respiratory tract infections, on average, than other persons at work and in his family. He has also been bothered by mild diarrheal episodes for most of his life. He has had a few urinary tract infections. On one occasion, a physician ordered a stool culture and stool examination for ova and parasites, and the culture was negative for stool pathogens, but Giardia lamblia cysts were identified in the stool smear. A CBC shows:
-
| Hgb |
14.2 g/dL |
| Hct |
42.6.2% |
| MCV |
90 fL |
| Platelet ct |
281,000/microliter |
| WBC count |
9860/microliter |
| WBC diff |
70 segs, 4 bands, 20 lymphs, 6 monos |
- He is involved in a motor vehicle accident and sustains major trauma to his lower legs, with significant blood loss. In hospital, he receives a blood transfusion. Soon after the transfusion is started, he develops severe hypotension.
- What do these findings suggest?
- The mild diarrhea and mild recurrent respiratory and urinary tract infections, along with the anaphylactic transfusion reaction, suggest an inherited deficiency of IgA.
- What laboratory testing would confirm this suspicion?
- Serum quantitative immunoglobulins show:
-
| IgA |
ND |
(16 - 83 mg/dL) |
| IgG |
816 mg/dL |
(282 - 1026 mg/dL) |
| IgM |
122 mg/dL |
(39 - 142 mg/dL) |
- Explain the pathophysiology of this condition:
- The major immunoglobulin elaborated by plasma cells in epithelia, mainly lamina propria, of the gastrointestinal tract and the respiratory tract is IgA. The IgA is present in the glycocalyx of the epithelial cells of the GI tract and in the mucociliary region of the respiratory tract, where it can neutralize organisms that attempt to pass through. There are high levels of the cytokine TGF-beta, which stimulates isotype switching to IgA. There is a subset of B cells in the gut that produce IgA in response to non-protein antigens typical for bacteria. Mucosal cells have Fc receptors that aid in transport of IgA through the cell to the lumenal surface. In addition, there are specialized microfold, or "M" cells, that overlie collection of gut-associated lymphoid tissue such as Peyer's patches and assist in the transfer of antigenic material to lymphocytes and macrophages.
- Persons who have inherited selective IgA deficiency have either failure in final differentiation of IgA-secreting B cells into plasma cells or decreased survival of plasma cells Normal numbers of T and B cells are present, but IgA secreting plasma cells are absent. In addition, deficiencies in IgG subclasses 2 and/or 4, while IgG subclasses 1 and 3 are increased in this disorder. Affected persons have an increased susceptibility to respiratory, urinary tract, and gastrointestinal infections (bacterial, and Giardia lamblia); mild diarrhea; half of cases have systemic anaphylaxis with blood transfusion from the presence of circulating antibodies to IgA.
- Case Presentation:
- History: A 12 year old boy is brought in by his mother. She has become increasingly concerned, because she is frequently being called by the school nurse who states that he is sick and needs to be taken home. This seems to be happening several times per month. Sometimes the problem is respiratory, while other times he seems to have a watery diarrhea. Past history reveals that, prior to age 5, he always seemed to be sick "about half the time" but the illnesses were minor and consisted of ear aches and runny nose. He was a term infant of normal birth weight, and the pregnancy was non-complicated. His sister, age 10, seems very healthy by comparison. The boy's father is known to have selective IgA deficiency (bothered only by occasional mild diarrhea). The boy's most recent respiratory infection is due to Streptococcus pneumoniae. A stool culture is negative for bacterial pathogens, but a stool for ova and parasites reveals Giardia lamblia cysts present.
- Physical examination: His height is 140 cm and his weight is 32 kg. Vital signs reveal temperature 37.6 C, respirations 25, pulse 82, and blood pressure 90/55. The oral mucosa is non-erythematous and the tympanic membranes are clear. The HEENT exam is otherwise unremarkable. Adenopathy is noted in cervical, axillary, and inguinal regions, but the nodes are non-tender. The heart rate is regular and no murmurs are audible. Examination of the chest reveals basilar rales on the left with dullness to percussion. Bowel sounds are present. No abdominal tenderness or masses are noted. The stool is guaiac negative. Motor strength is good in all extremities. Pulses are present and full. The skin is clear, except for some 1 to 4 cm diameter healing abrasions below the knees and on the left thigh. The neurologic exam is unremarkable.
- Laboratory findings:
-
| Antinuclear antibody (ANA) |
none detected |
| Nitroblue tetrazolium |
Unstimulated, 5% |
| MCV |
Stimulated, 81% |
-
| Sodium |
145 meq/L |
136 - 144 |
| Potassium |
4.9 meq/L |
3.7 - 5.2 |
| Chloride |
106 meq/L |
101 - 111 |
| CO2 |
23 meq/L |
20 - 29 |
| Urea Nitrogen (BUN) |
11 mg/dl |
7 - 20 |
| Creatinine |
0.6 mg/dl |
0.8 - 1.4 |
| Glucose |
92 mg/dl |
64 - 128 |
| Alk Phos |
325 U/L |
180 - 700 |
| LDH |
310 U/L |
105 - 230 |
| Creatine kinase |
75 U/L |
72 - 367 |
| AST |
32 U/L |
9 - 55 |
| ALT |
41 U/L |
3 - 46 |
| GGT |
20 U/L |
0 - 24 |
| Uric Acid |
2.9 mg/dl |
2.7 - 6.6 |
| Calcium |
9.2 mg/dl |
8.8 - 11.0 |
| Phosphorus |
4.5 mg/dl |
3.6 - 5.6 |
| Total Protein |
4.7 g/dl |
6.3 - 7.9 |
| Albumin |
4.5 g/dl |
3.9 - 5.0 |
| Globulin |
0.2 g/dl |
2.4 - 2.9 |
| Bilirubin, total |
0.5 mg/dl |
0 - 1.5 |
| Bilirubin, direct |
0.2 mg/dl |
0 - 0.3 |
| Cholesterol |
110 mg/dl |
100 - 200 |
- Quantitative Immunoglobulins
-
| IgA |
22 mg/dl |
68 - 378 |
| IgG |
175 mg/dl |
768 - 1632 |
| IgM |
40 mg/dl |
60 - 263 |
- Quantitative IgG subclasses
-
| IgG 1 |
132 mg/dl |
380 - 1420 |
| IgG 2 |
45 mg/dl |
73 - 455 |
| IgG 3 |
10 mg/dl |
16 - 194 |
| IgG 4 |
16 mg/dl |
1 - 153 |
- Complement, total (CH50)
-
- CBC
-
| WBC count |
7200/microliter |
3700 - 10,100 |
| Manual differential count |
55 segs, 2 bands, 35 lymphs, 6 monos, 2 eos |
| Hgb |
14.4 g/dl |
14.1 - 17.5 |
| Hct |
44.1% |
43.1 - 51.5 |
| MCV |
97 fL |
83 - 97 |
| Platelets |
295,000/microlter |
140 - 440 |
- Lymphocyte subsets
-
| CD4 cells (absolute) |
630 |
100 - 810 |
| CD8 cells (absolute) |
785 |
180 - 850 |
| B cells |
280 |
50 - 500 |
| T cells |
2010 |
820 - 2400 |
- Questions for Discussion:
- What, if anything, suggests an immunologic disease? Are there other conditions that could account for these findings?
Although young children may seem to be sick about half the time, the problems are usually minor. This child clearly has had many serious infections. Children with malignancies or congenital anomalies may also have a history of infections.
- What physical examination findings deserve attention?
He is below ideal body weight. He has lymphadenopathy. The rales (crackles) suggest an infiltrative process such as a pneumonia. A few abrasions are to be expected in any active child.
- What laboratory tests, if any, would be helpful?
Specimens can be obtained (sputum) to look for infection. In this case, the child's most recent respiratory infection is due to Streptococcus pneumoniae. A stool culture is negative for bacterial pathogens, but a stool for ova and parasites reveals Giardia lamblia cysts are present. Quantitative immunoglobulins and T and B cell counts are helpful in trying to distinguish the various forms of immunodeficiency (see laboratory findings).
- What radiologic procedures, if any, would be helpful?
A chest radiograph may help to diagnose a pneumonia.
- Based upon the findings, including the laboratory testing, what is the most likely diagnosis?
Although most patients with common variable immunodeficiency (CVID) present at an older age, there is a wide age range at presentation. The major feature is hypogammaglobulinemia, often involving all immunoglobulin classes but sometimes just IgG. The genetic defect in both selective IgA deficiency and CVID may be similar, and both conditions can be seen in the same family, and patients may present with either at a younger age in such a case. In most cases there are normal numbers of B cells, but they cannot readily differentiate into plasma cells. There may be deficient T cell regulation of B cell activity.
- Explain the pathophysiology of this condition.
This condition illustrates that the nature of the underlying problem can be complex. CVID is a disorder that does not always appear in children, which sets it apart from most primary immunodeficiency conditions. Half of CVID cases are diagnosed before age 21, but in some cases complications do not develop until adolescence or adulthood. Persons with CVID are prone to recurrent bacterial infections, particularly sinusitis, bronchitis, pneumonia, bronchiectasis, and otitis. Bordatella pertussis infections occur in childhood. Viral infections are uncommon, though recurrent herpes simplex with eventual herpes zoster is an exception. Giardiasis is common.
Persons with CVID have an increased incidence of autoimmune diseases, particularly hemolytic anemia, thrombocytopenia, and pernicious anemia. There is an increased tendency for nonspecific noncaseating sarcoid-like granulomas to be present in liver, spleen, lymph nodes, and bone marrow. Both Hodgkin's disease and non-Hodgkin's lymphomas are more frequent. Inflammatory bowel diseases are more common, and there is an increased risk for gastric carcinoma. Some affected persons develop amyloidosis.
In about two thirds of cases, normal numbers of circulating B lymphocytes are present. There is a decrease in immunoglobulins, generally in all classes, more often IgG and IgA, but sometimes only of IgG. At least two of the three main serum immunoglobulin isotypes are decreased. Diverse mechanisms, including failure of B cell maturation to plasma cells, excessive T cell suppression, or defective T helper cell function, may underlie CVID. A selective abnormality of T cell activation, as demonstrated by decreased synthesis of interleukins (IL 2, 4, and 5) has been identified in some cases. Other cases have T and B cell autoantibodies, or have a decreased CD4/CD8 ratio. CVID may be linked to selective IgA deficiency.
|
 Return to the Immunology Course Outline
Return to the Immunology Course Outline