


|
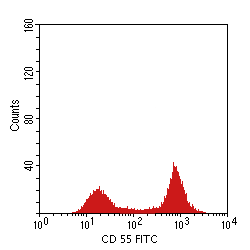
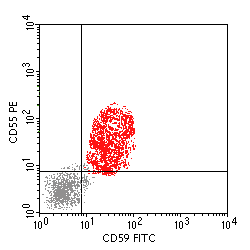
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired clonal disease of bone marrow stem cells, and all hematopoietic elements produced in the marrow, including hematopoiesis, myelopoiesis, and megakaryopoiesis, can be affeced. Since normal clones of stem cells do not completely disappear with PNH, the proportion of abnormal cells is variable among patients and can even vary over time in a single patient. PNH occurs when an inactivating somatic mutation in a gene on the X-chromosome occurs. This gene, known as pig-A or PIGA, is required for the biosynthesis of the glycosylphosphatidylinositol (GPI) anchor that attaches many proteins to the external cell membrane surface. A partial or complete absence of PIGA results in the absence of those proteins. Two such cell surface proteins that have functional significance are PNH decay accelerating factor (DAF), also known as CD55, and CD59. CD55 and CD59 block cell surface complement activation. When these proteins are deficient or absent, then RBCs are much more susceptible to complement-mediated lysis. In addition, platelets are more likely to initiate thrombosis. The most specific confirmatory testing for PNH involves flow cytometry to determine populations of peripheral blood cells with CD55 and CD59 binding, as shown above. In PNH, there will be a dual population of cells, one of which includes increased numbers of cells lacking CD55 and CD59. The proportion of abnormal cells to normal cells indicates the probable severity of the disease. The clinical manifestations of PHN relate to involvement of the three major hematopoietic cell lines: hemolytic anemia, thrombosis, granulocytopenia, and risk for leukemia. |
|
|