|
The API
QSTAR™ Pulsar Hybrid LC/MS/MS System is a high performance hybrid
quadrupole time-of-flight mass spectrometer designed for protein
identification and characterization. The system generates high quality MS
and MS/ MS data from both electrospray ionization (ESI) and
matrix-assisted laser desorption ionization (MALDI) techniques. The MS/
MS data can generate sequence tags of peptides, complete sequence of
peptides and can be used for the characterization of post translational
modifications. ESI-MS like all other mass spectrometric techniques is
based on the principle of producing molecular ions for subsequent
separation and analysis. ESI produces ions directly from liquid at
atmospheric pressure. For ESI measurements the samples solution are
infused into a glass capillary at a constant flow rate and introduced to
a "source", where intact ionized molecules in the gas phase are
produced. In the mass analyzer the molecular ions are separated on the
basis of their mass and charge. Unlike MALDI ions that usually carry a
charge of +1, ions generated by an ESI source usually carry several
charges, enabling better sequencing in MS/ MS modes.

ESI-QqTOF
Electrospray ionization
encompasses three different processes: droplet formation, droplet
shrinkage, and gaseous ion formation. At the onset of the electrospray
process, the electrostatic force on the liquid causes it to emerge from
the tip of the capillary as a jet in the shape of a "Taylor cone".
A thin liquid extends from this cone, which breaks into a mist of fine
droplets. Several factors such as the applied potential, the flow rate of
the solvent, the diameter of the capillary and solvent characteristics
influence the diameter of the initially formed droplets.
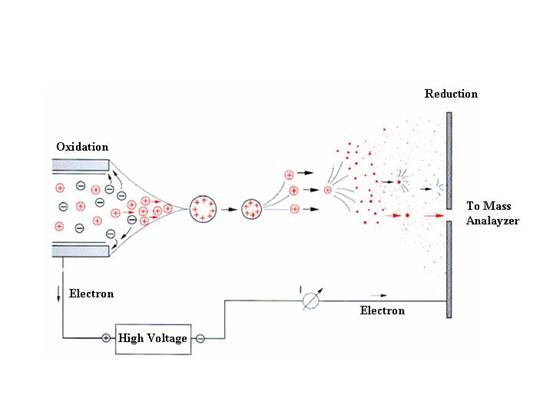
A key feature of the ESI
process is the formation of a series of multiply charged ions for large
biopolymers. The positive ions of the general nature [M+nH]n+ are formed by the
protonation of basic sites in biopolymers. In the negative ion mode, the
[M-nH]n- - type ions are
formed. The charge state of each ion can be used to determine the actual
mass instead of just the m/z ratio. The basis for this ability derives
from the fact that there are two common isotopes of carbon in nature ([12C]
and [13C] - with the ratio of [12C]/[13C] being about 99:1). As the peptide
gets larger it will contain more carbon atoms and hence there will be
increased probability it will contain two or more mass peaks for the same
peptide. That is, one mass peak for the peptide ion that only contains [12C]
isotopes and another mass peak (at m/z = +1) for a peptide ion molecule
that by chance happens to contain a single [13C] at one of its
carbon atoms). If the m/z difference is 0.5 the ion must be doubly
charged. Similarly, a difference of 0.333 or 0.25 corresponds with triply
and quadruply charged ions respectively.
Because a mass spectrometer
analyzes ions on the basis of their m/z ratios rather than their masses,
the effect of multiply charging is to reduce significantly the m/z of the
intact macromolecule, a process that brings high mass compounds within
the usable mass range of an ordinary mass spectrometer.
Peptide Sequencing
by Tandem Mass Spectrometry (MS/ MS)
Tandem mass spectrometry (MS/
MS) refers to the coupling of two mass spectrometers in time and space
with the objective to obtain further information of a more specific
nature about the sample in question. It takes advantage of the
fragmentation reactions that occur in the field free region of
multi-sector instruments. The concept of tandem mass spectrometry
involves mass selection, fragmentation, and mass analysis. The first
stage performs the mass selection of a specified ion from a mixture of
ions that are produced in the ion source. In the QSTAR the mass-selected
ion undergoes fragmentation in the intermediate region (Q2), via
collision with nitrogen or argon gas. The second stage of MS/ MS is used
to mass-analyze the product ions that are formed by the collision.
The most
promising ions for obtaining sequence data by electrospray mass
spectrometry are doubly charged trypric ions. In that case the charges
are located at the ends of the peptide, one at the N-terminus, and the
other at the C-terminal lysine or arginine. Thus, fragmentation results
in singly charge daughter ions. The cleavage leads to fragments of type an,
bn and cn if the charge is located at the end
terminus and to fragment of type xn, yn and zn
if the charge is retained on the C-terminus as shown in the next figure.
If a complete ion series is obtained, the analyzed peptide can be
sequence, because the adjacent signals differ by the mass of one amino
acid residue.

The isobaric amino acid
leucine and isoleucine and the modified amino acid hydroxyproline can not
be differentiated using a low energy collision-induced dissociation, such
as that of the QSTAR. High energy collisions produce an additional
fragments (dn and wn ions) allowing differentiation
between leucine and isoleucine.
The other pairs of isobaric
amino acid residues are lysine/glutamine and oxidated
methionine/phenylalanine. These two amino acids pairs can be
distinguished with high resolution instruments and accurate mass
measurements of fragment ions since in contrast to leucine and isoleucine
the exact masses of these amino acids residues are different (lysine
128.095, glutamine 128.059 and oxidated methionine 147.035, phenylalanine
147.068). Another possibility to distinguish between lysine and glutamine
is by acetylation of the peptide with acetic anhydrite. The acetylation
increases the mass by 42 Da for the free N-terminus and for every e-amino
group of lysine.
Relevant
literature
- Chapman, J. R., Mass Spectrometry of
Proteins and Peptides, 2001,Humana Press
- Dass, C., Principles and practice of
biological mass spectrometry, 2001, John Wiley & Sons
- James, P., Proteome research: mass
spectrometry, 2001, Springer
- Kellner, R., F. Lottspeich, and H. E. Meyer,
Microcharacterization of Proteins, 2nd Ed, 1999, Wiley-VCH.
- Kinter, M., and N. E. Sherman, Protein
Sequencing and Identification Using Tandem Mass Spectrometry, 2000,
Wiley Interscience
- Siuzdak, G., Mass Spectrometry for
Biotechnology, 1996, Academic Press
|