|
|
         |
Laboratory Guide: Inon Sharony, Tel:640-7634, room 411A, Ornstein Building. Welcome to the fascinating world of Molecular Dynamics!The following is a sample trajectory of 216 Lennard-Jones atoms (simulating liquid Argon) starting from a low entropy state and equilibrating to 90 K, calculated by the students as part of the experiment. Note that Periodic Boundary Conditions (PBC) have been enforced, such that an atom (labeled in red, starting from rear-bottom-left corner of the cell) which exits through one of the cell walls (in this case the rear wall) re-appears from the other side of the cell (i.e. the front wall). Ignoring the rest of the atoms, the motion of the labeled atom appears random.The movie was created from the output *.XYZ file using UCSF Chimera 1.6.1, then converted to a *.PNG image sequence using OpenShot, and then to a *.GIF format animation using GIMP.
Computational SimulationA computer simulation is a computer program used to simulate an abstract mathematical model of a particular system. Computer aided solutions to mathematical models are sometimes necessary when the mathematical model is proven to have no closed-form solution (i.e. it is too complex to be solved analytically), or if an analytical solution is just not practical (i.e. the resources required for an analytical solution, as opposed to a numerical one, are more than we would like to spend)."But [computers] are useless. They can only give you answers." -- Pablo Picasso, 1964.We will focus on how to get these answers and how to interpret them.Mathematical modelingIn constructing the model, abstraction is the key. As put forward from Aristotle to Einstein: "Everything should be made as simple as possible, but not simpler." In some contexts, this is referred to as Occam's razor. Computer simulations can introduce various levels of complexity, accordingly requiring increasing amounts of computational resources, from iteration by hand to distributed and High Performance Computing. Experiments conducted on a computer are sometimes whimsically termed "in silico".
Solving numericallyProblems may be modeled using various mathematical formulations, including (but not limited to) root finding & eigenvalue problems, differentiation & integration, probability & statistics, and more. Each formulation may have multiple numerical methods for its solution.
Molecular Dynamics
Molecular dynamics (MD) is a microscopic method that enables one to estimate thermodynamic variables defined by ensemble averages. It is based on a numerical solution of Newton's equations under the assumption of the validity of classical mechanics. Why go classical?One can raise the
question: "Why use classical mechanics when it seems Quantum dynamics govern the world?"
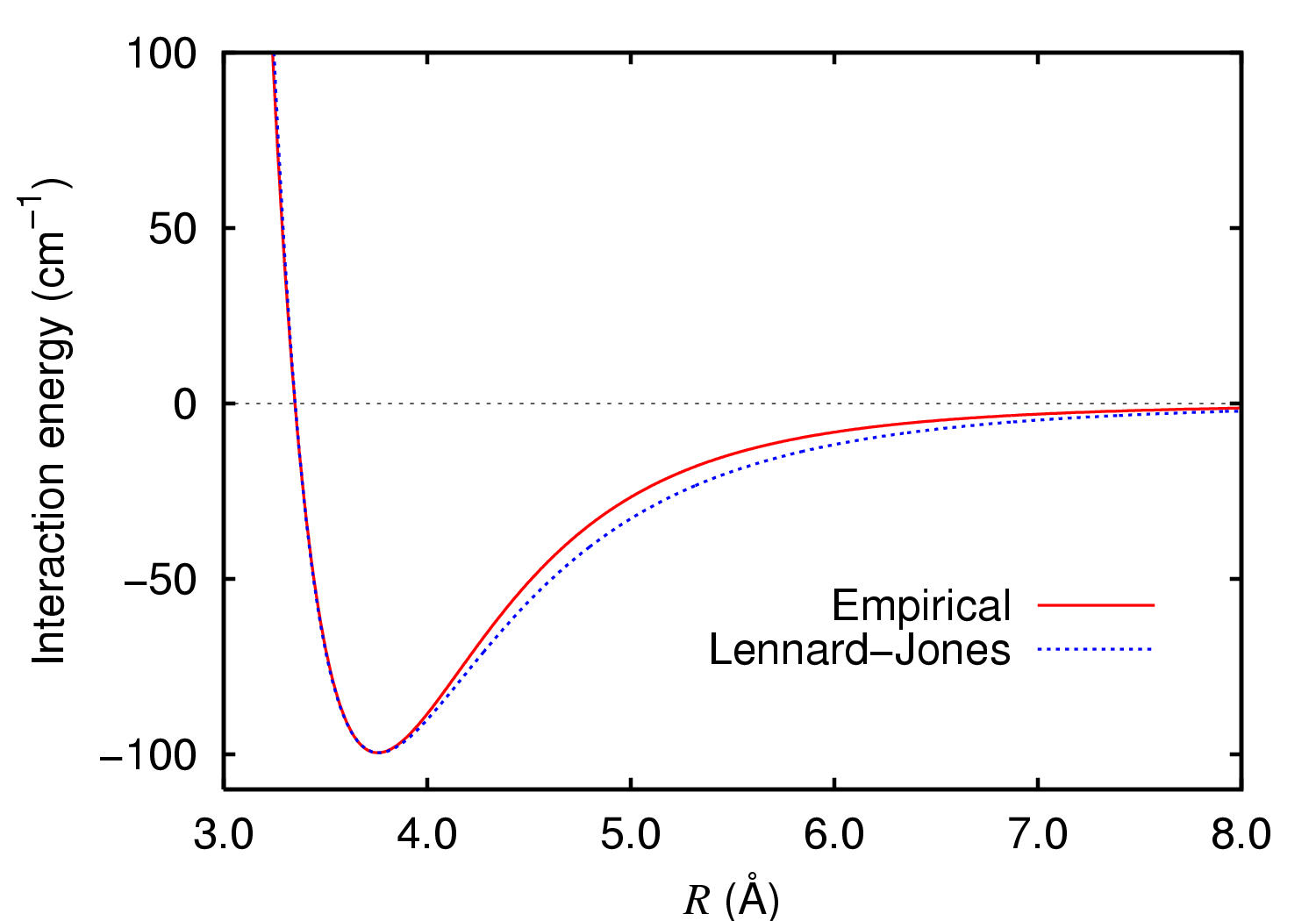
Choosing the potential surfaceThere are many potential surfaces that describe inter and intra molecular interactions. The choice of the potential surface has to reflect the general physical properties of the problem at hand. Common properties such as the restriction of atoms from occupying the same space (such as to reflect the Coulomb and Pauli repulsion) and vanishing interactions at large distances characterize many of existing models.Calibrating the parameters
|