
|
Research Interests |
Electronic, Magnetic, Mechanical, and Chemical Properties of Nanomaterials
The application of state-of-the-art Density Functional Theory calculations to the prediction and characterization of the electronic, magnetic, and mechanical properties of systems at the nanoscale is a major part of the research performed in our group. The following are some examples of research projects in this field that are performed in our group:
|
|
|
|
|
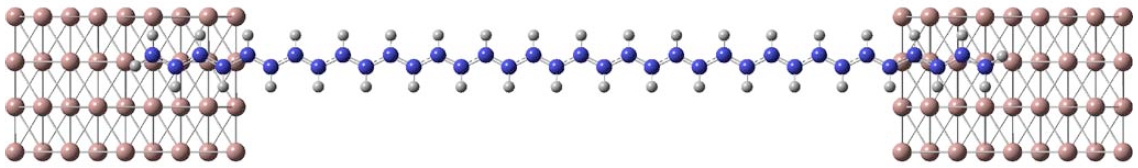
Electronic Transport through Nanoscale Constrictions
A unique method for efficient calculations of the electronic transport properties of finite elongated nanoscale systems is being developed in our group. Based on density functional theory combined with a carefully designed divide-and-conquer computational approach, we have been able to predict the electronic properties and transport through micrometer long molecules such as trans-polyacetylene, carbon nanotubes, and graphene nanoribbons. Further development to include finite two-dimensional structures is being pursued.
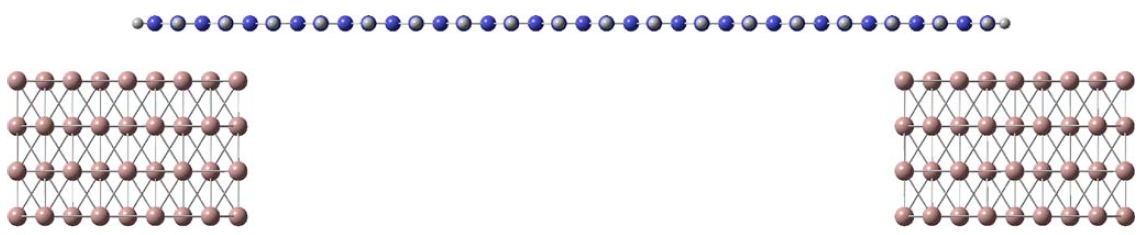 |
 |
Electron and Spin Dynamics in open systems
Time-domain simulation of electron dynamics in open systems is important for studying transient effects and transport mechanisms in molecular electronic devices. Based on time-dependent density-functional theory, a code is being developed, where efficient representation of the open system allows for the real-time simulation of electron dynamics in realistic molecular junction geometries.