7. ANALYSIS IN FLOW SYSTEMS
Exp. 1. Determination of nitrates in drinking water
In
a flow injection analysis the solution is pumped to the detector by a
peristaltic pump. Several tubes can be handled simultaneously on the same pump.
This feature permits to avoid the preliminary preparation of solutions of
analyte and reagent needed for a specific determination, and so high analysis
rate (about 100 samples per hour) can be reached.
The sample and all reagents are introduced from separate channels through the same pump or through different pumps. Complete mixing is achieved before the combined solution reacts the detector. Constant flow through each channel is of paramount importance for stable proportioning of sample to reagents.
Different modes of mixing the solutions of analyte and reagent are available. One of them, when two pumps are used, is shown on Fig.7-1.
Note: Peristaltic pump - device in which fluid is squeezed through plastic
tubing by rollers.
The flow rate is controlled by the speed of the motor (greater than 30
rpm) and the inner diameter of the microbore tubing (0.2 - 3 mm).
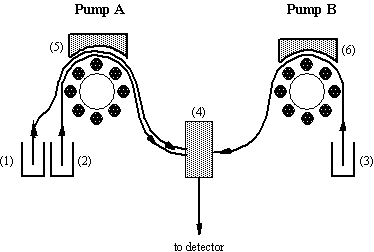
Fig.7-1 Scheme
of a flow system with two peristaltic pumps.
A timer
controls the time of operation of pump A. Solutions (1) and (2) are injected
via pump A, mixed in chamber (4) and flown to the detector. At the end of a
predetermined injection time pump A stops and pump B starts to transfer
solution (3) as long as needed up to next injection.
Preparation of
the flow system
·
Do not operate
the system without the assistance of the instructor.
·
Insert the
tubes into the solutions before turning on the pumps. If this is not done, the
air will penetrate into the tubes, distorting the smoothness of the flow.
·
Turn the pumps
off before changing solutions.
1. Set the plastic tubing on the
peristaltic pumps as shown on Fig.7-1. The pressure on plates (5) and (6) is
adjusted with the respective screws by the instructor.
2. Ensure that the polarographic detector
(Fig.7-2) is connected to the flow system.
3. Test for proper operation of the flow
system (use distilled water):
![]() Check that there is free flow
through each one of the three tubes. The flow may be obstructed due to blocking
of the thin tubes.
Check that there is free flow
through each one of the three tubes. The flow may be obstructed due to blocking
of the thin tubes.
![]() Check that solution from pump A
does not penetrate in tube (3). To perform the test, take out tube (3) from
beaker (3) while pump A is operating. No drops should be formed at that end of
the tube. Explain when such leakage is possible.
Check that solution from pump A
does not penetrate in tube (3). To perform the test, take out tube (3) from
beaker (3) while pump A is operating. No drops should be formed at that end of
the tube. Explain when such leakage is possible.
Repeat
the same test with pump B and tubes (1) and (2).
Preparation of
the polarographic detector
1.
Check that the container for drainage of mercury and waste is in place.
2.
Raise the level of the mercury reservoir and check that the mercury is
flowing.
3.
Nitrogen gas: Make sure that the fine needle valve, positioned on the
polarographic stand, is closed. Open the main nitrogen valve and adjust the
local pressure to about 0.4 bar. Turn on the peristaltic pump (use distilled
water with a flow rate about 0.5 - 1 ml/min). In absence of nitrogen air
bubbles are interlaced along the liquid flown through the degassing tube.
Note: BE AWARE! Mercury vapors are poisonous!
Notify the instructor in the event of mercury spill. Mercury should be
cleaned up immediately. Do not throw it down the drain.

Fig.7-2 High-performance
polarographic cell designed by Ch. Yarnitzky1.
The solution
is forced in the capillary tube by a peristaltic pump. In absence of nitrogen
air bubbles are interlaced along the degassing tube. When nitrogen gas
introduced, the solution is pushed to the walls of the capillary and forms a
thin film. The intimate solution-gas contact thus formed enables an efficient
purging of oxygen. The oxygen-free solution slides from the end of degassing
tube to the mercury capillary and is trapped by surface tension forces in the
gap between the end of the mercury capillary and the ceramic separator. This
gap comprises the entire electrochemical cell: the mercury electrode from
above, the counter electrode from below and the reference Ag/AgCl/1 M KCl
electrode at the end of the lower tube. The volume of the cavity is about 0.1
cm3.
Carefully
open the fine needle valve (positioned on the polarographic stand) and observe
the flow of the liquid across the tube. The nitrogen gas pushes the liquid
against the walls of the tube, and an invisible thin liquid film is formed.
Increase the nitrogen flow up to the stage, at which the thin film is stable,
as marked by a seemingly motionless state along the tube. Observe the flow of
the liquid along the mercury capillary and in the cell cavity. It is smooth and
steady. These are the right operating conditions. A further increase of the
nitrogen flow distorts the smoothness of the flow in the cell cavity.
Chemicals 1. 0.1 M KCl
2. 0.2 mM UO2 (CH3COO)2,
0.02 M HCl, 0.2 M KCl
3. 5 mM KNO3
Procedure
1.
Prepare six standard solutions of KNO3 in the
concentration range of 0.05 mM - 0.3 mM (0.05, 0.10, 0.15, 0.20, 0.25, 0.30
mM). You are provided with six 50 ml volumetric flasks and six plastic beakers,
each marked with the respective concentration. The beakers are used for
aspirating the standard solutions and the samples.
2.
The principle of the experiment is demonstrated by
recording DC polarograms in each of the following three solutions:
I. Nitrate and KCl
(as supporting electrolyte).
II. Uranyl acetate
and KCl (as supporting electrolyte).
III. Nitrate with
uranyl acetate (as catalyzer) and KCl (as supporting electrolyte).
What do you expect
to obtain in each one of the three cases in respect to limiting currents and E1/2?
The
arrangement of the solutions for this experiment is shown in Fig.7-3. Only pump
A is used, and the timer is set to a long enough time to permit the recording
of a whole polarogram. The advised order for recording the polarograms is III,
II, I. Record the three polarograms at the same sensitivity. What are your
conclusions from the above polarograms?
Determine
the value of the potential corresponding to the limiting current of the
catalyzed nitrate wave. At this potential the current-time curves of the
samples and the standard solutions, flown through the detector, are recorded.
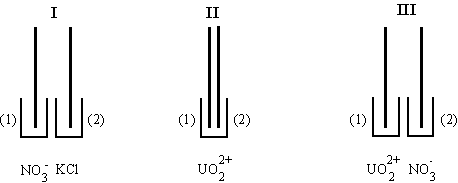
Fig.7-3 Arrangement
of solutions for demonstrating the principle of the nitrate determination.
Only pump A is used. Sets I, II and III -
different experimental configurations of solutions.
UO22+ - solution of
uranyl acetate with HCl and KCl;
NO3-
- solution of nitrate ion; KCl - supporting electrolyte.
3.
The analytical determination of nitrates. Set the
potential to the value corresponding to the limiting current of the catalyzed
nitrate wave.
The
arrangement of the solutions is given in Fig.7-4. Tube (1) is dipped in the
same uranyl acetate solution throughout the entire analysis. The samples and
the standard solutions are aspirated from tube (2). For building the
calibration curve inject each of the standard solutions and record the current
signal. The injection time is controlled by pump A (20 sec). At the end of this
time pump B starts to operate as long as needed for rinsing the detector.
Repeat several times the injection of the analyte, record the current and
collect for each solution at least 3 - 4 signals with good reproducibility.
Plot the calibration curve.
Repeat
the procedure with the drinking water samples. If the signal is higher than
that of the most concentrated standard, dilute the analyte (why?). Calculate
the concentration of nitrates in the sample.
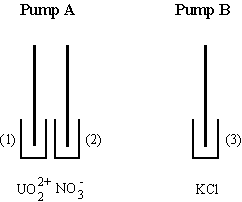
Fig.7-4 Arrangement of solutions for the routine analysis of nitrates. Both
pumps are used. Pump A injects the sample or standard solution of nitrates and
the make up solution of uranyl acetate for a predetermined time. Pump B
transfers the supporting electrolyte as long as needed.
4. At the end of the experimental
session:
·
Rinse the
system (tubing and cell cavity) by passing distilled water for about two
minutes.
·
Release the
tension applied on the peristaltic pump tubes.
·
Lower the
mercury reservoir.
·
Nitrogen gas: Close
the fine needle valve located on the polarographic stand and turn off the main
nitrogen valve.
Reference
1. M.Noufi, Ch.Yarnitzky and M.Ariel, Anal.
Chim. Acta, 1990, 228, 117.
Exp. 2. Determination of fluoride in drinking water with ion-selective electrode
Fluoride
ion-selective electrode
Electrodes
employing ion-selective membranes can display high selectivity toward certain
ions. No electrode, however, responds exclusively to one kind of ion. The
sensitivity of an electrode to a competing ion, N, is characterized by the
selectivity coefficient kM,N, defined at equal concentrations of M
and N.
kM,N =
response to M / response to N
The
response of an electrode specific to an ion M in presence of a competing ion N
is:
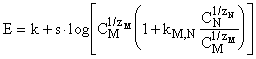
where CM
and CN are the concentrations of ions M and N, z is the charge of
ion. The value s is in the range 54 - 60 mV/decade at 25°C and at constant ionic strength.
The
fluoride ion-selective electrode is a solid-state electrode employing a crystal
of LaF3, doped with Eu(II). The doping of the crystal enables the
fluoride ion to migrate throughout the membrane.
The
response of the fluoride electrode in the presence of a competing anion N is
given by equation:

where CF
and CN are the concentrations of fluoride and of the competing
species N.
The
fluoride electrode yields a nearly Nernstian response over a concentration
range of 1 - 10-6 M F- at a constant ionic strength and
at a constant temperature.
The
electrode operates with relatively few interferences. Interfering species are:
·
hydronium
ions, which below pH 5 form HF and HF2- complexes;
·
iron(III),
silicon, aluminum and other polyvalent cations, which also form complexes with
fluoride ion;
·
hydroxide
ions, which compete with the fluorides with a selectivity coefficient kF,OH
= 0.1.
The
degree of interference caused by the competing ion OH- is determined by the term kF,OH·COH / CF:
for
kF,OH·COH / CF << 1,
the interference is negligible;
for
kF,OH·COH / CF >>1,
the effect of the competing ion is predominant.
The
effect of interference at low-level concentrations of fluoride at different pH
values is given in table below. In order to ensure negligible effect of the OH-
competing ion, the pH should be lower than 7.
|
Degree of interference of OH-, expressed as kF,OH·COH / CF |
||
|
pH |
kF,OH·COH / CF |
|
|
|
CF
= 10-5 M |
CF
= 10-6 M |
|
11 |
10 |
100 |
|
10 |
1 |
10 |
|
9 |
0.1 |
1 |
|
8 |
0.01 |
0.1 |
|
7 |
0.001 |
0.01 |
The
time response of the electrode (the time required to reach 99% of the stable potential
reading) varies from several seconds in concentrated solutions to several
minutes near the limit of detection.
Measurements
with a fluoride electrode are performed in presence of TISAB (Total Ionic
Strength Adjusting Buffer). TISAB should provide constant ionic strength of the
solution, adjusting the pH and complex the interfering species.
The
type of TISAB depends on the composition of the sample. Drinking water contains
less than 1.5 ppm F- (7.5·10-5
M) and negligible amounts of interfering species. A suitable composition of
TISAB for drinking water is 0.01 M acetate buffer pH 5 - 5.5 in 1 M NaCl and 4
g/l CDTA. The TISAB is added to the sample in volume ratio 1:1. The acetate
buffer keeps the pH at the optimal value, at which the formation of HF and HF2-
is negligible, and the response of the electrode to hydroxides is virtually
zero. NaCl is added to maintain constant ionic strength, independent of the
composition of the sample. For the specific configuration of the reference
electrode (AgCl-coated silver wire), used in this experiment, the constant
concentration of chlorides is needed to ensure the constant value of the
electrode potential. CDTA, chelating agent similar to EDTA, complexes
polyvalent ions, which otherwise complex with F-.
The effect of
Fe(III)-F complexes on the determination of fluorides in drinking water
Fe(III)
forms three complexes1 with F-: FeF2+, FeF2+,
FeF3.
|
Fe3+
+ F- = FeF2+ |
Kfl =
105 |
|
FeF2+ + F- = FeF2+ |
Kf2 =
103.9 |
|
FeF2+
+ F- = FeF3 |
Kf3 =
103 |
Only uncomplexed,
free fluoride is measured by the fluoride selective electrode. In order to
illustrate the effect of complexation, the concentration of the different
complexes as function of the total concentration of F- is shown in
Fig.7-5(a). The calculations are performed for 2·10-5 M Fe(III), concentration considered as the highest
permissible in drinking water. The concentration of uncomplexed iron decreases
with increasing concentration of fluoride. Around [F-]total
= 5·10-5 M the predominant complex is
FeF2+. Beyond [F-]total = 1·10-4 M the predominant species is
FeF2+.
The
data from Fig.7-5(a) are also presented as ![]() , the fraction of free fluoride to the total concentration of
all fluoride species, as a function of the total concentration of fluoride
(Fig.7-5(b)). The value of
, the fraction of free fluoride to the total concentration of
all fluoride species, as a function of the total concentration of fluoride
(Fig.7-5(b)). The value of ![]() is considerably
lower than unity in the typical concentration range of fluoride in drinking
water. The lower the total concentration of Fe3+, the higher
is considerably
lower than unity in the typical concentration range of fluoride in drinking
water. The lower the total concentration of Fe3+, the higher ![]() .
.
For
the determination of the total F- concentration, the chelating agent
CDTA (forming a complex with Fe(III) with a high formation constant) is added.
This allows the determination of F- concentration, independent of
the level or nature of dissolved minerals. In a 1 ppm fluoride sample the TISAB
complexes about 5 ppm iron or aluminum.


Fig.7-5 Concentration of the Fe(III)-F complexes (a) and ![]() (b) as
function of the total concentration of F-.The calculations are
performed for [Fe3+ ]total = 2·10-5 M. The dashed area
represents the concentration range of fluorides, typical to drinking water.
(b) as
function of the total concentration of F-.The calculations are
performed for [Fe3+ ]total = 2·10-5 M. The dashed area
represents the concentration range of fluorides, typical to drinking water.
Experimental
setup
The
experiment can be performed in a batch mode or in a flow system. The flow
system shown in Fig.7-6 is highly efficient, saving reagents and time of
operator.
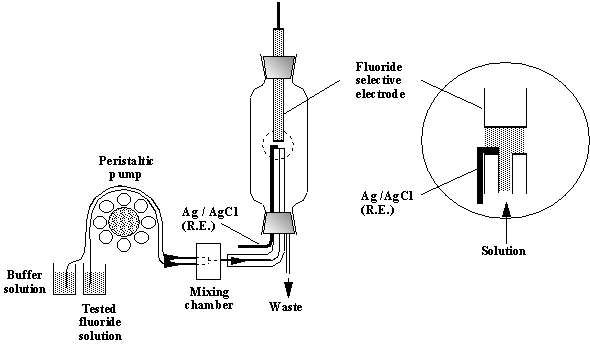
Fig.7-6 A flow system for determination of fluorides.
Chemicals 1. 10-2 M NaF
2. 10-3 M NaF
3. TISAB containing 0.01 M acetate buffer
pH 5-5.5, 1 M NaCl and 4 g/l
CDTA
(1,2-diaminocyclohexane N,N,N',N'-tetraacetic acid)
Electrodes: Fluoride
ion-selective electrode and Ag/AgCl reference electrode (silver wire coated
with AgCl)
Procedure
Check
the connection of the electrodes to the pH-meter. Set the meter to voltage
reading.
Calibration
curve for the fluoride standards is constructed in the range 10-2 -
5·10-6 M. Prepare standard
solutions of 10-4 M, 3·10-5
M, 10-5 M and 5·10-6 M NaF
in 50 ml volumetric flasks. Transfer each of the fluoride solutions (standards
and analytes) and the TISAB via the flow system (cf. Fig.7-5) at a flow rate of
5 to 10 ml/min. It is recommended to perform the measurements in decreasing
order of concentrations. Record stable voltage reading of the potential between
the electrodes for each fluoride solution. One minute or more should be allowed
to reach steady state readings, depending on the concentration of F-.
The lower the concentration, the longer the time.
At
the end of the working session rinse the system with distilled water.
Construct
a calibration curve (potential vs concentration of fluoride). Mark the
X-axis in mole/l and ppm. Report the concentration of the fluorides in drinking
water samples in mole/l and ppm.
Reference
1. “Stability constants of metal-ion
complexes”, L. G. Sillen and A. E. Martell, Special Publication #17, London:
The Chemical Society, Burlington House, 1964.
Recommended
Literature
Exp. 1.
Determination of nitrates in drinking water
1. H.
H. Willard, L. L. Merritt, J. A. Dean and F. A. Settle, Instrumental Methods
of Analysis.
2. D.
A. Skoog and D. M. West, Principles of Instrumental Analysis.
3. D.
A. Skoog and J. J. Leary, Instrumental Analysis.
4. I.
M. Kolthoff, W. E. Harris and G. Matsuyama, J. Am. Chem. Soc., 66
(1944), 1782.
5. H.
Hemmi, K. Hasebe, K. Ohzeki and T. Kambara, Talanta, 31 (1984),
319.
6. M.
Noufi, Ch. Yarnitzky and M. Ariel, Anal. Chim. Acta, 234
(1990), 475.
Exp. 2. Determination
of fluorides in drinking water
1. D.
C. Harris, Quantitative Chemical Analysis.
Go to Main Page